一种氮硼双掺杂的螺环化合物及其制备方法和应用
1.本发明涉及有机电致发光材料技术领域,具体涉及一种氮硼双掺杂的螺环化合物及其制备方法和应用。
背景技术:
2.螺环化合物是由两个分子片段通过一个sp3杂化的碳原子连接而成的,由于螺环骨架的空间效应,使前线分子轨道可以充分分离。两个分子片段在螺环结构中垂直排列可以抑制非辐射跃迁,避免严重的聚集浓度猝灭,刚性的螺环结构可以实现高效的空间电荷转移和高的光致发光量子效率。螺环化合物具有独特的三维结构,展现出良好的热稳定性和形貌稳定性。因此,螺环化合物受到广大科研工作者的青睐,成为最重要的一类有机半导体材料。
3.热激活延迟荧光是三重态激子的一种热激活再发光的过程,即三重态热激活后转化到更高的振动能级,再通过反向系间窜越到达与其能级接近的单重态的振动能级,通过辐射跃迁产生延迟荧光,寿命通常在μs量级。相对应的,单重态激子从单重态迅速衰减到基态直接辐射发光为瞬时荧光,寿命在ns量级。热激活延迟荧光材料是继传统荧光材料和贵金属磷光材料之后发展起来的第三代纯有机发光材料。热激活延迟荧光材料不仅具备传统荧光材料和磷光材料的优越性,而且避免了重金属参与的不足,近年来受到广泛关注。
4.近年来,蒋佐权教授等人提出基于螺环结构的空间限制电荷转移概念,通过这种机制制备出给体-受体-给体型的π堆叠高效有机发光材料。王悦教授将螺环电子给体和螺环电子受体通过大位阻基团相连,构筑了具有热激活延迟荧光性质的高性能深蓝光电致发光材料。显然,螺环化合物由于其独特的几何和电子结构,在热活化延迟荧光型发光材料领域具有很大的应用潜力。然而,通过给体和受体之间的相互作用来调节材料的发光性能及其构效关系还尚未系统研究。
5.硼的空p轨道与邻近的π-共轭体系之间的p-π*共轭作用,大幅降低π-共轭体系的 lumo能级,从而赋予其独特的光电性能。基于这些特点,科研人员已开发出许多具有热激活延迟荧光性质的含硼多环芳烃。但是,将硼原子引入到螺环主体骨架并利用含硼多环芳烃的受体性质来调节热激活延迟荧光材料的发光性能的研究极少。
技术实现要素:
6.为了克服现有技术的上述缺点与不足,发明人设计合成了一系列硼、氮双掺杂的螺环分子,通过对给体、受体单元的有效调控,开发出具有热激活延迟荧光性质的有机小分子发光材料。本发明提供的氮硼双掺杂的螺环分子具有高的荧光量子效率,良好的溶解性和热稳定性,将其作为发光层通过溶液法制备的oled器件开启电压低,最大外量子效率高达22%,体现了其在oled发光材料领域的应用潜力。
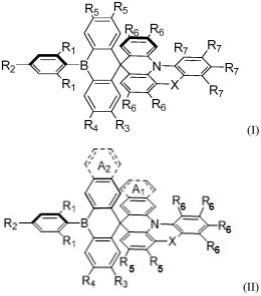
7.本发明第一个目的是提供一种氮硼双掺杂的螺环化合物,具有如下式(i)或式(ii)所示的化学结构:
[0008][0009]
在通式(i),(ii)中r1、r2、r3、r4、r5、r6和r7选自如下一种或多种取代基:氢原子、卤素原子、含有1-4个碳原子的烷基、卤代烷基、硝基、磺酸基、氰基、甲酰基、乙烯基、烷酰基和烷基硫化物基团;x为o、s、n或c(r8)2,其中r8为含有1-4个碳原子的烷基,或者x不存在;
[0010]
式(ii)化合物中,环a1,a2独立地为*-cra=cr
a-cra=cr
a-*,ra选自如下一种或多种取代基:氢原子、卤素原子、含有1-4个碳原子的烷基、卤代烷基、硝基、磺酸基、氰基、甲酰基、乙烯基、烷酰基和烷基硫化物基团和苯基稠合的位点;或者环a1,环a2中的一个不存在;
[0011]
式(i)和式(ii)的化合物中,r1和r2不同时为h。
[0012]
进一步地,所述卤素原子选自f、cl、br或i;所述含有1-4个碳原子的烷基选自甲基、乙基、正丙基、异丙基、正丁基或叔丁基;所述卤代烷基选自cf3、ccl3、cbr3、ch2f、 chf2、ch2cf3或cf2cf3。
[0013]
氮硼双掺杂的螺环化合物为以下结构:
[0014]
[0015][0016]
本发明第二个目的是提供一种上述氮硼双掺杂的螺环化合物的制备方法,合成路线如下:
[0017][0018]
上述化合物的制备方法,包括以下步骤:
[0019]
式(i)化合物的制备:在惰性气氛下,中间体a
′
的溶液,反应体系冷却至-80℃~-60℃的低温,加入正丁基锂,保持低温搅拌1-2h,缓慢加入中间体b
′
的溶液,滴加完毕后缓慢升温至室温,搅拌过夜,后处理后,加入冰乙酸和盐酸,加热回流反应5-10h,提纯得到化合物(i)。
[0020]
式(ii)化合物的制备:在惰性气氛下,中间体a
″
的溶液,反应体系冷却至-80℃~-60℃的低温,加入正丁基锂,保持低温搅拌1-2h,缓慢加入中间体b
″
的溶液,滴加完毕后缓慢升温至室温,搅拌过夜,后处理后,加入冰乙酸和盐酸,加热回流反应5-10h,提纯得到化合物(ii)。
[0021]
进一步地,中间体a
′
或a
″
的溶液,中间体b
′
或b
″
的溶液中所用溶剂为四氢呋喃、乙醚中的至少一种;所述缓慢升温是以2-3℃/min的升温速率升温;所述后处理是猝灭反应 (比如加入饱和氯化铵溶液),水相用溶剂萃取(溶剂为二氯甲烷、氯仿和乙酸乙酯等),有机相洗涤(饱和氯化钠溶液洗涤),干燥(常规干燥剂处理,比如无水硫酸钠),旋蒸除去溶剂。
所述提纯的方法为本领域所熟知,比如重结晶,或者柱层析提纯。
[0022]
进一步地,中间体a
′
(或者中间体a
″
)、正丁基锂、中间体b
′
(或者中间体b
″
)的摩尔比为1:1-1.2:1-1.2,优选地,正丁基锂,中间体b
′
(或者中间体b
″
)稍稍过量;进一步地,冰乙酸的体积加入量是中间体a
′
(或者中间体a
″
)质量的50-60倍(ml/g),盐酸的体积加入量是中间体a质量的3-5倍(ml/g)。
[0023]
本发明第三个目的是提供一种有机电致发光器件,包括上述硼、氮双掺杂的螺环化合物作为发光层材料。
附图说明
[0024]
图1是硼、氮双掺杂的螺环化合物的紫外可见吸收光谱(图1a)和荧光发射光谱(图1b);
[0025]
图2是硼、氮双掺杂的螺环化合物的在不同溶剂中的发射光谱和荧光照片;
[0026]
图3是硼、氮双掺杂螺环化合物的循环伏安曲线;
[0027]
图4是硼、氮双掺杂的螺环化合物在不同溶剂中,分别在无氧和有氧条件下的的瞬态荧光光谱;
[0028]
图5是硼、氮双掺杂螺环化合物5wt%掺杂膜的发射光谱和瞬态荧光衰减曲线;
[0029]
图6是硼、氮双掺杂螺环化合物在掺杂膜中的变温瞬态荧光衰减曲线以及在掺杂膜中的 k
risc
与温度的关系;
[0030]
图7是硼、氮双掺杂螺环化合物的热重分析图;
[0031]
图8是硼、氮双掺杂螺环化合物作为发光层器件的电致发光光谱图;
[0032]
图9是硼、氮双掺杂螺环化合物作为发光层器件的电流密度-电压-发光亮度关系图;
[0033]
图10是硼、氮双掺杂螺环化合物作为发光层器件的外量子效率-发光亮度曲线;
[0034]
图11是硼、氮双掺杂螺环化合物作为发光层器件的电流密度-发光亮度曲线;
[0035]
图12是基于硼、氮双掺杂螺环化合物性能展示图。
具体实施方式
[0036]
制备例1
[0037]
先以中间体b
′
中r1、r2、r3为cf3,r4、r5为h的化合物,命名为fmesb-f
aq的制备方法如下:
[0038][0039]
(1)化合物1a的合成
[0040]
将1-溴-2-碘-4-(三氟甲基)苯(7.02g,20.0mmol)置于250ml反应瓶中,在真空线上抽换氮气3次,于氮气氛围下加入120ml干燥的四氢呋喃。将反应瓶放入到-15℃的低温仪中,随后向反应体系中缓慢滴加异丙基氯化镁氯化锂(1.3m in thf,16.9ml,22.0 mmol),反应液在-15℃下搅拌2小时。再将反应瓶置于-78℃环境中,用注射器缓慢滴加邻溴苯甲醛(4.07g,22.0mmol),滴加结束后缓慢升至室温搅拌过夜。用6m的hcl 淬灭反应,直到水层显示石蕊红色,再加入50ml水,水相用乙酸乙酯萃取3次,合并有机相并用饱和nacl溶液洗涤,有机相用无水na2so4干燥。旋蒸除去溶剂,粗品用柱层析纯化,洗脱剂为石油醚:乙酸乙酯=8:1,得到7.1g白色固体1a,产率为87%。
[0041]1h nmr(700mhz,cdcl3)δ7.79(s,1h),7.70(d,j=8.4hz,1h),7.61(d,j=7.7hz, 1h),7.46(d,j=8.4hz,1h),7.30(t,j=7.7hz,1h),7.21(t,j=8.4hz,1h),7.12(d,j=7.7 hz,1h),6.43(d,j=4.2hz,1h),2.66(d,j=4.2hz,1h).
13
c nmr(176mhz,cdcl3)δ 142.30,140.26,133.65,133.30,130.25(q,j=33.4hz),130.02,128.74,128.05,127.37,126.12 (q,j=3.5hz),125.62(q,j=3.5hz),124.36,123.93(q,j=272.8hz),74.08.[m+h
–
h2o]
+ calcd.for c
14h19
br2f3o,392.8924;found:392.8913.
[0042]
(2)化合物1b的合成
[0043]
将化合物1a(6.2g,15.1mmol)置于250ml圆底烧瓶中,加入120ml乙酸,再加入氢碘酸(9.7ml,57%in h2o,60.5mmol),随后将反应体系置于120℃的油浴中,加热回流反应2小时。待冷却至室温后,加入饱和的亚硫酸钠溶液淬灭反应,反应液由黑色变成深黄色。再加入100ml水稀释,用二氯甲烷萃取3次,将合并的有机相冷却到0℃,缓慢加入naoh水溶液(1m),直到呈石蕊蓝。有机相用饱和nacl溶液洗涤,并用无水 na2so4干燥。旋蒸除去溶剂,浓缩得到的液体用柱层析纯化,洗脱剂为石油醚,得到4.8g 无色液体1b,产率为80%。
[0044]1h nmr(700mhz,cdcl3)δ7.73(d,j=8.4hz,1h),7.62(d,j=7.7hz,1h),7.37(d,j =8.4hz,1h),7.26-7.23(m,2h),7.15(t,j=7.7hz,1h),6.98(d,j=7.7hz,1h),4.24(s,2h). 13
c nmr(176mhz,cdcl3)δ140.20,137.85,133.56,133.24,130.75,130.19(q,j=33.4hz), 129.09,128.70,127.87,127.39(q,j=3.5hz),125.14,124.96(q,j=3.5hz),123.88(q,j=272.8hz),42.23.hr-esims(m/z):[m-h]+calcd.for c
14
h9br2f3,390.8950;found: 390.8967.
[0045]
(3)化合物1c的合成
干燥。旋蒸除去溶剂,粗品用柱层析纯化,洗脱剂为石油醚:二氯甲烷=1:1,得到0.64g浅黄色固体fmesb-f
aq,产率为72%。
[0053]1h nmr(400mhz,cd2cl2)δ8.66(s,1h),8.44(d,j=8.0hz,1h),8.30(s,2h),7.83(t,j =8.0hz,2h),7.65(t,j=8.0hz,1h),7.43(d,j=8.0hz,1h),7.32(d,j=8.0hz,1h).
13
c nmr(176mhz,cd2cl2)δ186.51,142.18(br,b-c),140.19(br,b-c),139.22,138.41,137.69, 137.49,136.93(br,b-c),136.13(q,j=33.4),134.66(q,j=33.4),134.06,133.03(q,j=35.2), 129.75,129.72,128.82,128.55,126.98,125.33(q,j=3.5),124.03(q,j=274.6),123.93(q,j= 272.8),123.19(q,j=272.8).
11
b nmr(225mhz,cd2cl2)δ60.7.hr-esims(m/z):[m+h]
+ calcd.for c
23
h9bf
12
o,541.0628;found:541.0625.
[0054]
制备例2
[0055]
以中间体b
″
中r1、r2为cf3,r3、r4为h,a2环为苯环的化合物,命名为fmes-tq,制备方法写下:
[0056][0057]
(1)化合物2a的合成
[0058]
将2,3-二溴萘(2.86g,10.0mmol)置于250ml反应瓶中,在真空线上抽换氮气3次,于氮气氛围下加入70ml干燥的四氢呋喃。将反应瓶置于-15℃的低温仪中,随后向反应体系中缓慢滴加异丙基氯化镁氯化锂(1.3m in thf,8.46ml,11.0mmol),反应液在-15℃下搅拌2小时。再将反应瓶置于-78℃环境中,用注射器缓慢滴加甲酸乙酯(0.41ml,5.05 mmol),滴加结束后缓慢升至室温搅拌过夜。用6m的hcl淬灭反应,直到水层显示石蕊红色,再加入50ml水,水相用乙酸乙酯萃取3次,合并有机相并用饱和nacl溶液洗涤,有机相用无水na2so4干燥。旋蒸除去溶剂,粗品用柱层析纯化,洗脱剂为石油醚:乙酸乙酯=10:1,得到3.0g白色固体2a,产率为68%。
[0059]1h nmr(400mhz,cdcl3)δ8.13(s,2h),7.81(s,2h),7.77(t,j=8.0hz,4h),7.54
‑ꢀ
7.47(m,4h),6.69(s,1h),2.75(s,1h).
13
c nmr(101mhz,cdcl3)δ138.47,134.08,132.30, 131.88,128.41,128.13,127.25,126.79,126.73,121.57,74.50.
[0060]
(2)化合物2b的合成
[0061]
将化合物2a(3.0g,7.65mmol)置于圆底烧瓶中,加入60ml乙酸作溶剂,再加入氢碘酸(4.0ml,57%in h2o,30.6mmol),用锡箔纸包裹反应装置,随后将反应体系置于 120℃的油浴中,加热回流反应2小时。待冷却至室温后,加入饱和的亚硫酸钠溶液淬灭反应,反应液由黑色变成深黄色。再加入80ml水稀释,用二氯甲烷萃取3次,将合并的有机相冷却到0℃,缓慢加入naoh水溶液(1m),直到呈石蕊蓝。有机相用饱和nacl 溶液洗涤,并用无水na2so4干
燥。旋蒸除去溶剂,浓缩得到的液体用柱层析纯化,洗脱剂为石油醚,得到1.73白色固体2b,产率为60%。
[0062]1h nmr(400mhz,cdcl3)δ8.14(s,1h),7.77-7.74(m,1h),7.70-7.66(m,1h),7.65 (d,j=8.0hz,1h),7.49-7.43(m,2h),7.41(s,1h),7.24(t,j=8.0hz,1h),7.15(t,j=8.0hz, 1h),7.02(d,j=8.0hz,1h),4.35(s,2h).
13
c nmr(176mhz,cdcl3)δ139.12,136.35, 133.43,133.04,132.56,131.55,130.95,129.41,128.30,127.70,126.74,126.61,126.53,125.28, 123.36,42.47.
[0063]
(3)化合物2c的合成
[0064]
将化合物2b(1.5g,3.98mmol)置于反应瓶中,在真空线上抽换氮气3次,于氮气氛围下加入50ml干燥的四氢呋喃。将反应瓶放入到-78℃的低温仪中,随后向反应体系中缓慢滴加正丁基锂(1.6m;5.3ml,8.4mmol),反应液在-78℃下搅拌1.5小时。用注射器滴加二甲二氯化锡(0.97g,4.4mmol)的四氢呋喃(2ml)溶液,滴加结束后缓慢升至室温搅拌过夜。加入饱和氯化铵溶液淬灭反应,水相用乙酸乙酯萃取3次,合并有机相并用饱和nacl溶液洗涤,有机相用无水na2so4干燥。旋蒸除去溶剂得到黄色油状物质,用 c18反相填料柱层析纯化,洗脱剂为乙腈,得到无色液体,在0℃保存后,凝固成1.0g 白色固体2c,产率为70%。
[0065]1h nmr(400mhz,cdcl3)δ8.04(s,1h),7.80(s,1h),7.79-7.76(m,2h),7.58(d,j= 8.0hz,1h),7.45-7.40(m,3h),7.30-7.20(m,2h),4.15(s,2h),0.62(s,6h).
13
c nmr(101 mhz,cdcl3)δ147.16,143.29,141.02,139.65,136.35,136.05,133.96,132.02,129.02,128.37, 127.62,127.34,126.20,126.10,125.64,125.51,46.84,-9.94.hr-esims(m/z):[m+h]
+
calcd. for c
19h19
sn,367.0530;found:367.0499.
[0066]
(4)化合物fmesb-t的合成
[0067]
将化合物2c(1.0g,2.74mmol)置于100ml schlenk瓶中,在真空线上抽换氮气3次,于氮气氛围下加入30ml无水二氯甲烷。将反应瓶放入到-78℃的低温仪中,随后向反应体系中逐滴加入三氯化硼(1.0m;4.11ml,4.11mmol),滴加完毕后缓慢升至室温搅拌过夜。次日用真空泵将反应体系中的溶剂除去,再将反应瓶置于60℃的油浴中,在动态真空环境下去除残留的me2sncl2,留下灰色固体2d(0.61g,85%),该样品对水分和氧气敏感,因此未进行进一步提纯而直接用于后续反应。在100ml schlenk瓶中加入fmes(0.98 g,3.48mmol),真空线上抽换氮气3次,加入60ml无水乙醚,将反应瓶置于-78℃的低温仪中,随后向反应体系中缓慢滴加正丁基锂(1.6m;2.4ml,3.83mmol),滴加完毕后在-78℃搅拌0.5小时,随后升至室温搅拌4小时。用真空泵除去反应体系中的乙醚,得到黄色固体(fmes锂盐),往该反应瓶中加入15ml无水甲苯,并置于-78℃环境中,再向其中加入化合物2d的甲苯溶液,加完后自然升至室温搅拌过夜。次日加入30ml水淬灭反应,水相用二氯甲烷萃取3次,有机相用饱和nacl溶液洗涤,并用无水na2so4干燥。旋蒸除去溶剂,浓缩后的粗品用柱层析纯化,洗脱剂为石油醚,得到0.53g黄色固体fmesb-t,产率为45%。
[0068]1h nmr(400mhz,cd2cl2)δ8.30(s,2h),8.04(s,1h),7.92-7.89(m,2h),7.80(d,j= 8.0hz,1h),7.64(d,j=4.0hz,2h),7.59(t,j=8.0hz,1h),7.45(t,j=8.0hz,1h),7.36(d,j =8.0hz,1h),7.32-7.28(m,1h),4.75(s,2h).
13
c nmr(101mhz,cd2cl2)δ148.27,141.93, 140.01,137.62,136.38,134.78(q,j=32.3hz),133.96,131.95,131.91(q,j=34.3hz),129.52, 128.95,128.65,127.59,126.65,126.25,126.24,125.91,124.21(q,j=
276.7hz),123.53(q,j= 272.7hz),38.34.
11
b nmr(128mhz,cd2cl2)δ58.7.hr-esims(m/z):[m+h]
+
calcd.for c
26h14
bf9,509.1118;found:509.1042.
[0069]
(5)化合物fmesb-tq的合成
[0070]
将化合物fmesb-t(0.50g,0.98mmol)和三氧化铬(0.26g,2.55mmol)置于圆底烧瓶中,加入35ml乙酸,随后将反应体系置于120℃的油浴中,加热回流反应12小时。冷却至室温后,加入50ml水,用二氯甲烷萃取3次,合并有机相并用饱和nahco3溶液洗3次,用无水na2so4干燥。旋蒸除去溶剂,粗品用柱层析纯化,洗脱剂为石油醚:二氯甲烷=1:1,得到0.37g浅黄色固体fmesb-tq,产率为72%。
[0071]1h nmr(400mhz,cdcl3)δ8.69(s,1h),8.30(d,j=8.0hz,1h),8.27(s,2h),8.03(d,j =8hz,1h),7.90(s,1h),7.83(d,j=8hz,1h),7.75-7.68(m,2h),7.63(t,j=8.0hz,1h), 7.57(t,j=8.0hz,1h),7.43(d,j=8hz,1h).
13
c nmr(101mhz,cdcl3)δ187.74,140.46, 139.56,137.03,136.13,135.13,134.90,134.85(q,j=32.3hz),134.11,133.30,132.46(q,j= 34.3hz),130.44,130.40,129.69,129.66,129.05,128.64,126.34,123.72(q,j=276.7hz), 122.96(q,j=273.7hz).
11
b nmr(128mhz,cdcl3)δ59.8.hr-esims(m/z):[m+h]
+
calcd. for c
26h12
bf9o,523.0910;found:523.0923.
[0072]
制备例3
[0073]
化合物npa-br的合成(中间体a的合成参照此方法)
[0074][0075]
将二苯胺(0.5g,2.95mmol),2,3-二溴萘(1.01g,3.55mmol),叔丁醇钠(0.43g,4.43 mmol),三(二亚苄基丙酮)二钯(0.082g,0.09mmol)和三叔丁基膦(0.043g,0.21mmol) 置于100ml反应瓶中,加入30ml无水甲苯,将反应瓶置于液氮中冷冻15分钟,再用油泵抽真空10分钟,排除反应瓶中的空气,再将体系解冻,按照上述操作步骤重复3次。将反应瓶置于100℃油浴中加热12小时。冷却至室温后,加入20ml水,用二氯甲烷萃取3次,合并有机相并用饱和nacl溶液洗3次,用无水na2so4干燥。旋蒸除去溶剂,粗品用柱层析纯化,洗脱剂为石油醚:二氯甲烷=8:1,得到0.46g白色固体,产率为42%。
[0076]1h nmr(400mhz,cd2cl2)δ8.22(s,1h),7.80(t,j=4.0hz,1h),7.73-7.69(m,2h), 7.52-7.46(m,2h),7.24(t,j=8.0hz,4h),7.02-6.96(m,6h).
13
c nmr(176mhz,cd2cl2) δ147.81,143.35,133.89,133.76,133.10,130.31,129.45,127.69,127.08,127.05,122.82, 122.46,122.41.hr-esims(m/z):[m+h]
+
calcd.for c
22h16
brn,374.0539;found:374.0521.
[0077]
实施例1
[0078]
化合物tpa-s-mes*b(i-5)的合成
[0079][0080]
将2-溴-三苯胺(0.33g,1.0mmol)置于50ml schlenk瓶中,在真空线上抽换氮气3 次,于氮气氛围下加入10ml无水四氢呋喃,再将反应体系置于-78℃的低温仪中,随后向反应体系中缓慢滴加正丁基锂(1.6m;0.69ml,1.1mmol),反应液在-78℃下搅拌1.5 小时。用注射器滴加mes*b-aq(0.52g,1.2mmol)的四氢呋喃(5ml)溶液,滴加结束后缓慢升至室温搅拌过夜。加入饱和氯化铵溶液淬灭反应,水相用二氯甲烷萃取3次,合并有机相并用饱和nacl溶液洗涤,有机相用无水na2so4干燥。将有机相置于单口瓶中,旋蒸去溶剂,加入20ml冰乙酸和1ml盐酸,加热回流反应6小时。冷去至室温后,加入30ml水,用布氏漏斗过滤,滤渣用柱层析纯化,洗脱剂为石油醚:二氯甲烷=3:1,得到0.43g白色固体,产率为65%。
[0081]1h nmr(400mhz,cd2cl2)δ7.78-7.74(m,2h),7.67(dd,j1=7.6hz,j2=1.6hz,2h), 7.64-7.60(m,1h),7.58(s,2h),7.54(d,j=8.0hz,4h),7.42-7.37(m,2h),7.18(t,j=8.0 hz,2h),6.90-6.84(m,2h),6.55-6.50(m,4h),6.35(d,j=8.0hz,2h),1.45(s,9h),1.27(s, 18h).
13
c nmr(176mhz,cd2cl2)δ158.18,153.10,148.93,141.49,140.16,138.13,137.45, 133.97,132.85,132.63,131.63,131.46,131.25,128.95,127.12,126.13,123.28,120.59,115.36, 53.35,38.90,35.51,35.02,31.57.
11
b nmr(225mhz,cd2cl2)δ62.6.hr-esims(m/z):[m+ h]
+
calcd.for c
49h50
bn,664.4109;found:664.4127.
[0082]
实施例2
[0083]
化合物phx-s-mes*b(i-6)的合成
[0084][0085]
化合物phx-s-mes*b的合成与化合物tpa-s-mes*b的合成方法一致,得到0.33g白色固体,产率为54%。
[0086]1h nmr(400mhz,cd2cl2)δ7.73-7.07(m,16h),6.76(t,j=8.0hz,2h),6.63(t,j=8.0hz,1h),6.53(d,j=8.0hz,1h),6.11(d,j=8.0hz,1h),1.44(s,9h),1.24(s,9h),1.21(s, 9h).
13
c nmr(176mhz,cd2cl2)δ155.68,154.34,152.97,152.94,149.12,149.05,147.15, 140.98,138.15,137.12,136.75,135.20,134.69,134.05,133.82,133.49,132.38,131.81,131.60, 129.05,127.07,126.61,125.28,124.25,123.51,123.26,
123.23,123.12,117.77,116.98,116.44, 114.19,53.19,38.90,35.44,35.03,31.57.
11
b nmr(225mhz,cd2cl2)δ62.1.hr-esims (m/z):[m]
+
calcd.for c
49h48
bno,677.3829;found:677.3814.
[0087]
实施例3
[0088]
化合物tpa-s-f
mesb(i-13)的合成
[0089][0090]
化合物tpa-s-f
mesb的合成与化合物tpa-s-mes*b的合成方法一致,得到0.35g黄色固体,产率为50%。
[0091]1h nmr(400mhz,cd2cl2)δ8.33(s,2h),7.76(t,j=8.0hz,2h),7.62(t,j=8.0hz, 1h),7.56-7.48(m,6h),7.29(d,j=8.0hz,2h),7.21-7.17(m,2h),6.89-6.84(m,2h), 6.55(t,j=8.0hz,2h),6.42(d,j=8.0hz,2h),6.34(d,j=8.0hz,2h).
13
c nmr(176mhz, cd2cl2)δ161.11,145.78,141.44,139.91,136.58,134.89,134.78(q,j=31.9hz),133.00, 131.99(q,j=33.4hz),131.68,131.65,131.43,129.00,128.67,127.20,126.05,124.31(q,j= 274.6hz),123.46(q,j=272.8hz),120.89,115.53,53.27.
11
b nmr(225mhz,cd2cl2)δ62.8. hr-esims(m/z):[m-h]
+
calcd.for c
40h23
bf9n,700.1853;found:700.1844.
[0092]
实施例4
[0093]
化合物tpa-s-f
mesbf(i-17)的合成
[0094][0095]
化合物tpa-s-f
mesbf的合成与化合物tpa-s-mes*b的合成方法一致,得到0.35g 黄色固体,产率为58%。
[0096]1h nmr(400mhz,cd2cl2)δ8.35(s,2h),7.86(s,1h),7.77(t,j=8.0hz,2h),7.63(t,j =8.0hz,1h),7.59-7.53(m,4h),7.42(s,2h),7.33(d,j=8.0hz,1h),7.23(t,j=8.0hz, 1h),6.93-6.88(m,2h),6.58(t,j=8.0hz,2h),6.41-6.38(m,4h).
13
c nmr(176mhz, cd2cl2)δ161.16,160.95,144.74,141.17,140.02,137.03,136.91,135.72,135.55,135.35, 134.81(q,j=31.7hz),133.06,132.39(q,j=35.2hz),131.72,131.55,131.21,129.08(q,j= 3.52hz),127.88,127.63,126.84,126.41,125.70,124.26(q,j=274.6hz),124.15,
124.14(q,j =272.8hz),122.60,122.55,122.54,122.52,121.12,115.78,54.24.
11
b nmr(225mhz, cd2cl2)δ59.3.hr-esims(m/z):[m+h]
+
calcd.for c
41h22
bf
12
n,768.1726;found: 768.1705.
[0097]
实施例5
[0098]
化合物phx-s-f
mesbf(i-18)的合成
[0099][0100]
化合物phx-s-f
mesbf的合成与化合物tpa-s-mes*b的合成方法一致,得到0.64g 黄色固体,产率为68%。
[0101]1h nmr(400mhz,cd2cl2)δ8.34(s,2h),7.75(d,j=8.0hz,1h),7.58-7.07(m,12h), 6.81-6.79(m,2h),6.68(t,j=4.0hz,1h),6.45(d,j=4.0hz,1h),6.05(d,j=4.0hz,1h). 13
c nmr(176mhz,cd2cl2)δ158.08,149.17,147.31,144.46,137.61,135.07,134.76(q,j=31.7hz),133.87,132.46(q,j=35.2hz),131.49,131.43,129.24,127.57,126.91,126.85, 126.57,126.25,125.01,124.89,124.70,124.49,124.35(q,j=272.8hz),124.30,124.21(q,j= 274.6hz),124.05,123.60,123.45,123.15,123.05,121.59,117.90,117.48,116.34,114.77, 53.47.
11
b nmr(225mhz,cd2cl2)δ60.5.hr-esims(m/z):[m]
+
calcd.for c
41h20
bf
12
no, 781.1446;found:781.1452.
[0102]
实施例6
[0103]
化合物npa-s-f
mesbf(ii-1)的合成
[0104][0105]
化合物npa-s-f
mesbf的合成与化合物tpa-s-mes*b的合成方法一致,得到0.23g 黄色固体,产率为56%。
[0106]1h nmr(400mhz,cdcl3)δ8.34(s,1h),8.32(s,1h),7.84-7.79(m,3h),7.67(t,j= 8.0hz,1h),7.59(d,j=8.0hz,2h),7.52(d,j=8.0hz,1h),7.47(t,j=8.0hz,1h),7.39(t,j =8.0hz,2h),7.36-7.27(m,3h),7.21(t,j=8.0hz,2h),7.08(t,j=8.0hz,1h),6.97-6.93 (m,2h),6.63(t,j=8.0hz,2h),6.49(d,j=8.0hz,1h),6.42(d,j=8.0hz,1h).
13
c nmr (176mhz,cdcl3)δ160.68,160.51,144.65,141.01,139.90,138.35,136.75,136.54,135.63, 135.42,134.67(q,j=31.7hz),133.90,133.03,133.00,132.28(q,j=33.4hz),
1h),6.12(d,j=8.0hz,1h).
13
c nmr(176mhz,cd2cl2)δ157.95,149.34,147.16,145.52, 137.08,135.94,134.95(q,j=31.7hz),133.91,132.14(q,j=33.4hz),132.13,131.67,129.29, 129.02,128.96,128.03,126.96,126.78,126.62,125.44,125.11,124.32(q,j=274.6hz), 124.29,123.82,123.53,123.50(q,j=272.8hz),123.46,121.97,117.92,117.35,116.20, 114.15,52.94.
11
b nmr(225mhz,cd2cl2)δ60.9.hr-esims(m/z):[m]
+
calcd.for c
44h23
bf9no,764.1802;found:764.1786.
[0117]
为了方便,将上述制备例制备得到氮硼双掺杂的螺环化合物,化合物(i-5)命名为tpa-s-mes*b,化合物(i-6)命名为phx-s-mes*b,化合物(i-13)命名为tpa-s-f
mesb,化合物(i-17)命名为tpa-s-f
mesbf,化合物(i-18)命名为phx-s-f
mesbf,化合物(ii-1)命名为 npa-s-f
mesbf,化合物(ii-2)命名为npa-s-f
mesbt,化合物(ii-3)命名为phx-s-f
mesbt。
[0118]
应用例
[0119]
下面对本实施例的氮硼双掺杂螺环双小分子发光材料进行测试
[0120]
(1)光物理性质分析
[0121]
测试了上述氮硼双掺杂的螺环化合物的紫外可见吸收光谱(溶剂二氯甲烷),如图1(a) 所示,位于230-310nm的强吸收带表明分子内存在n-π*和π-π*跃迁吸收,在310-370nm 出现弱的吸收带,归属于芳基胺到三芳基硼的电荷转移吸收,表明在基态电子结构中给体和受体之间存在强的电子耦合。螺环化合物的这种通过螺碳原子的相互作用可被归因于匀共轭(homoconjugation)效应。与其他化合物相比,化合物npa-s-f
mesbf,npa-s-f
mesbt 和phx-sfmesbt具有扩展的π共轭,进而导致其电荷转移吸收带发生明显红移。
[0122]
还测试了氮硼双掺杂的螺环化合物的荧光发射光谱(溶剂甲苯)。如图1(b)所示,可以看出以三苯胺为给体的化合物tpa-s-mes*b、tpa-s-f
mesb和tpa-s-f
mesbf的发射峰分别为459nm、511nm和555nm,并展现出较高的荧光量子效率(82-94%)。含吩噁嗪单元的化合物phx-s-mes*b、phx-s-f
mesbf和phx-s-f
mesbt的发射峰分别为482 nm、608nm和534nm,荧光量子效率分别为33%、58%和6%。npa-s-f
mesbf和 npa-s-f
mesbt的发射峰分别567nm和535nm,荧光量子效率仅为18%和2%。结果表明,含三苯胺的化合物在甲苯中的发光较强,而npa-s-f
mesbf、npa-s-f
mesbt和 phx-s-f
mesbt这些具有拓展π体系的分子在甲苯中的发光较弱。与tpa-s-mes*b相比, phx-s-f
mesbf在甲苯中的发射红移了149nm,这是由于含吩噁嗪的给体单元相比三苯胺基团具有更强的给电子能力,使得分子带隙变窄所致。
[0123]
为了进一步研究这些化合物的发光行为,我们测试了氮硼双掺杂螺环化合物在不同溶剂中的发射光谱。如图2所示,其中图2(a)为化合物tpa-s-mes*b的发射光谱和荧光照片,图2(b)为化合物phx-s-mes*b的发射光谱和荧光照片,图2(c)为化合物tpa-s-f
mesb的发射光谱和荧光照片,图2(d)为化合物tpa-s-f
mesbf的发射光谱和荧光照片,图2(e)为化合物phx-s-f
mesbf的发射光谱和荧光照片,氮硼双掺杂螺环化合物的发射光谱对溶剂极性具有依赖性随着溶剂极性的增加,这些化合物的发射峰逐渐红移,且发射峰逐渐变宽。以tpa-s-mes*b为例,其在正己烷中的发射峰位于426nm,在n,n-二甲基甲酰胺中的发射峰位于519nm,发射光谱红移了93nm并伴随着发光颜色从蓝色到绿色的变化。由此表明,bn掺杂螺环化合物具有分子内电荷转移特性,表现出从基态到激发态的较大跃迁偶极矩。
[0124]
(2)电化学性质分析
[0125]
为了研究氮硼双掺杂的螺环化合物的相对能级,采用循环伏安法对其进行了电化
学测试。如图3所示,为氮硼双掺杂的螺环化合物的循环伏安曲线,可以看出,这些化合物均表现出可逆的氧化还原峰,表明其具有良好的电化学稳定性。根据氧化还原起始电位可估算出这些化合物的homo/lumo能级,计算结果如表1所示。在mes*取代的化合物中,相同的受体单元使它们的lumo能级非常接近,由于含吩噁嗪单元的给电子能力强于三苯胺,因此phx-s-mes*b相对tpa-s-mes*b更容易被氧化。对于化合物tpa-s-mes*b、 tpa-s-f
mesb和tpa-s-f
mesbf,三个化合物具有相同的三苯胺给体单元导致它们的 homo能级相仿,而对于lumo能级,由于tpa-s-f
mesbf中具有更强的吸电子基团, tpa-s-f
mesbf表现出更低的lumo能级。phx-s-mes*b、phx-s-f
mesbf和 phx-s-f
mesbt具有相同的给体单元而呈现出相似的homo能级,phx-s-f
mesbf因其具有更强的吸电子基团使其更容易被还原。npa-s-f
mesbf和npa-s-f
mesbt则表现出相似的homo和lumo能级。结果如下表1所示:
[0126]
表1氮硼双掺杂的螺环化合物的光物理和电化学数据
[0127][0128]
注:a.在二氯甲烷中(1.0
×
10-5
m-1
)的吸收起峰值,b.氮气下甲苯中的发射峰和绝对荧光量子效率,c.氧化/还原峰的半波电位,d.e
lumo
/e
homo
=-(4.8+e
red
/e
ox
)
[0129]
(3)热活化延迟荧光性质分析
[0130]
测试了氮硼双掺杂螺环化合物在不同溶剂中的瞬态荧光衰减曲线,图4是化合物 tpa-s-mes*b、phx-s-mes*b、tpa-s-f
mesb、tpa-s-f
mesbf和phx-s-f
mesbf在不同溶剂中的瞬态荧光衰减曲线,左图是在无氧条件下测试,右图是在有氧条件下测试。图4(a) 是tpa-s-mes*b在不同溶剂中的瞬态荧光光谱;图4(b)是phx-s-mes*b在不同溶剂中的瞬态荧光光谱;图4(c)是tpa-s-f
mesb在不同溶剂中的瞬态荧光光谱;图4(d)是 tpa-s-f
mesbf在不同溶剂中的瞬态荧光光谱;图4(e)是phx-s-f
mesb在不同溶剂中的瞬态荧光光谱。可以看出,这些化合物在无氧条件下均表现出双指数衰减过程,其中包括纳秒时间尺度的瞬时荧光和微秒时间尺度的延迟荧光,表明它们在稀溶液中符合热激活延迟荧光(tadf)特征。当这些化合物的溶液暴露于空气后,再对其进行瞬态光谱测试,发现没有明显的延迟组分,表明这些化合物产生的激发三重态被氧气猝灭。
[0131]
为了进一步研究氮硼双掺杂的螺环化合物的tadf性质,测试了在掺杂薄膜中的光
物理性质,采用旋涂的方法制备了氮硼双掺杂的螺环化合物以5wt%掺杂在主体材料中的薄膜,其中tpa-s-mes*b和phx-s-mes*b掺杂在二[2-((氧代)二苯基膦基)苯基]醚(dpepo) 中,tpa-s-f
mesb、tpa-s-f
mesbf和phx-s-f
mesbf掺杂在4,4'-二(9-咔唑)联苯(cbp) 中。图5是硼、氮双掺杂螺环化合物5wt%掺杂膜的发射光谱和瞬态荧光衰减曲线。其中图5(a)是硼、氮双掺杂螺环化合物5wt%掺杂膜(tpa-s-mes*b、phx-s-mes*b掺杂在dpepo中,tpa-s-f
mesb、tpa-s-f
mesbf和phx-s-f
mesbf掺杂在cbp中)的发射光谱。mes*取代的化合物tpa-s-mes*b和phx-s-mes*b在掺杂膜中呈现深蓝色和蓝色发光,其荧光量子效率分别为65%和56%。fmes取代的化合物tpa-s-f
mesb、tpa-s-f
mesbf 在掺杂膜中分别呈现绿色和黄绿色光,其发射峰位分别在508nm和538nm,虽然这两个分子的主体骨架相同,但tpa-s-f
mesbf的硼杂蒽环多了一个吸电子取代基cf3,导致其在掺杂膜中的发射光谱红移30nm,这两个化合物在掺杂膜中的荧光量子效率高达100%。 phx-s-f
mesbf在掺杂膜中呈现黄色光,其发射峰在562nm,荧光量子效率为97%。值得一提的是,与甲苯溶液中的发射峰相比,掺杂膜中的发射峰发生蓝移,这可能与激发态条件下分子结构的重组和弛豫受到周围固体介质的限制以及化合物在掺杂薄膜中分子间相互作用减小有关。同样的,采用旋涂的方法制备了氮硼双掺杂的螺环化合物的纯薄膜,图 5(c)是氮硼双掺杂螺环化合物纯薄膜的发射光谱,含三苯胺的化合物tpa-s-mes*b、 tpa-s-f
mesb和tpa-s-f
mesbf在薄膜中的发射峰分别位于451nm、496nm和536nm,与对应的掺杂膜相比发生了蓝移,其荧光量子效率为22-58%,显著低于掺杂膜中的荧光量子效率,表明氮硼双掺杂的螺环化合物掺杂在主体材料中实现了有效的能量转移。在室温条件下,进一步测试了氮硼双掺杂螺环分子的薄膜在氮气氛围下的瞬态荧光衰减曲线(图 5(b)和5(d)),表2为氮硼双掺杂螺环化合物在薄膜中的光物理数据,掺杂膜和纯薄膜中的瞬态荧光衰减曲线均呈现出两个不同的组份:一个纳秒级的瞬变衰减和一个微秒级的延迟衰减,表明氮硼双掺杂螺环分子在薄膜中符合tadf特征。
[0132]
我们进一步测试了氮硼双掺杂的螺环化合物在掺杂膜中的变温瞬态荧光衰减曲线。图 6(a-e)是氮硼双掺杂螺环化合物在掺杂膜中的变温瞬态荧光衰减曲线,随着温度的升高,延迟荧光比例逐渐增加,表明延迟荧光是通过反隙间窜越过程产生的。图6(f-j)是氮硼双掺杂螺环化合物在掺杂膜中的k
risc
与温度的关系图,根据adachi等人报道的热活化能δe
st
的计算方法,k
risc
与δe
st
的关系为:k
risc
∝
aexp(-δe
st
/kbt),其中a是频率因子,kb和 t是玻尔兹曼常数和温度。基于掺杂膜中的光物理数据,根据adachi等人的方法,利用阿伦尼乌斯表达式,计算出这些分子的δe
st
。硼氮杂螺环分子在掺杂膜中的δe
st
为28.5-75.5 mev,均符合tadf材料的发光特征。
[0133]
表2硼氮杂螺环化合物在薄膜中的光物理数据
[0134][0135]
(4)热学性质分析
[0136]
在氮气氛围下对这些化合物进行了热重测试,如图7所示,氮硼双掺杂螺环化合物的热分解温度均在275.4℃以上。在含硼杂蒽单元的化合物中,mes*取代的化合物相比fmes 取代的化合物具有更高的热分解温度。此外,在fmes取代的化合物中,与含硼杂蒽的化合物相比,含硼杂并四苯的化合物表现出更高的热分解温度。这些结果表明,氮硼双掺杂螺环化合物均具有良好的热稳定性,这有利于化合物在制备光电器件时保持稳定性。
[0137]
(5)光电性能分析
[0138]
鉴于氮硼双掺杂螺环化合物的tadf特征和良好的热稳定性,我们以三苯胺为给体的分子(tpa-s-mes*b,tpa-s-f
mesb和tpa-s-f
mesbf)作为发光层,通过溶液法制备了电致发光器件。器件的结构为:ito/pedot:pss(30nm)/mcp:emission layer(50nm)/ tspo1(20nm)/tmpypb(20nm)/lif(1nm)/al(100nm),其中ito和al分别为阳极和阴极,pedot:pss(聚乙撑二氧噻吩-聚(苯乙烯磺酸盐))作为空穴注入层和空穴传输层, mcp(1,3-二咔唑-9-基苯)为主体材料,tspo1(二苯基[4-(三苯基硅烷基)苯基]氧膦)为空穴阻挡层,tmpypb(1,3,5-三(3-吡啶基-3-苯基)苯)和lif(氟化锂)分别为电子传输层和电子注入层。器件制备:首先,ito玻璃基底在洗涤剂、去离子水、丙酮和异丙醇依次超声清洗10分钟,放在120℃的烘箱烘烤5分钟。在旋涂pedot:pss薄膜之前,用等离子体处理20分钟,然后将pedot:pss溶液以1800转/分的转速旋转涂覆在ito 玻璃基片上,在120℃下退火10分钟。再将含有pedot:pss的基片转入充满氮气的手套箱中。将发光材料(30wt%掺杂于mcp)溶于氯苯,以2000转/分的转速旋转涂覆在 pedot:pss上面,在50℃退火10分钟,放入真空沉积室。在5
×
10-4
pa的压力下,依次蒸镀有机层、金属阴极,有机物以的速率、lif以金属铝以金属铝以的速率进行蒸镀。器件测试:器件的j-v-l曲线是在手套箱中使用keithley 2400源表测量。制备的器件有效面积为10mm2,均未封装。器件的电致发光(el)光谱由光学分析仪pr-745记录。器件的外量子效率通过驱动电流于相应的器件亮度和电致发光光谱计算得到(假定器件为朗伯光源)。
[0139]
图8是电致发光光谱和对应的器件照片及cie色标,图9是电流密度-电压-亮度曲线,图10是亮度-外量子效率曲线,图11是亮度-电流效率曲线,电致发光器件的数据如表3 所示。从图8可以看出,tpa-s-mes*b,tpa-s-f
mesb和tpa-s-f
mesbf的的发光峰位分别为454nm、502nm和536nm,这与对应化合物在掺杂膜中的光致发光光谱非常类似。 tpa-s-mes*b,tpa-s-f
mesb和tpa-s-f
mesbf的开启电压分别为2.6v、2.2v和3.0v,表明基于这些材料的器件具有较低的开启电压。
[0140]
表3基于氮硼双掺杂螺环化合物的器件相关数据
[0141][0142]
其中:a开启电压;b最大亮度;c最大电流效率;d最大功率效率;e最大外量子效率;f 100cd m-2
时的cie。
[0143]
器件性能测试表明,基于tpa-s-f
mesbf的器件最大亮度为4797cd m-2
,最大电流效率为77.38cd a-1
,最大功率效率为490.69lm w-1
,最大外量子效率为22%,这样的器件性能在目前报道的溶液法制备的基于螺环结构tadf器件的属于前列。这些结果表明,在氮硼双掺杂的螺环化合物中,通过增强受体单元的吸电子能力有助于提升器件的性能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1