稳定标记细菌新生蛋白的方法
1.本发明涉及蛋白标记技术领域,具体涉及一种稳定标记细菌新生蛋白的方法。
背景技术:
2.细菌向外界分泌蛋白,以满足自身生长,增殖及在环境内生存的需求。胞内病原菌通过自身的分泌系统向宿主细胞内注射效应蛋白(foultier b, troisfontaines p, m
ü
ller s, opperdoes fr, cornelis gr. characterization of the ysa pathogenicity locus in the chromosome of yersinia enterocolitica and phylogeny analysis of type iii secretion systems. journal of molecular evolution. 2002 jul;55(1):37-51.),使得细胞骨架重排,产生细胞因子等进而破坏宿主细胞功能(cornelis gr. the type iii secretion injectisome. nat rev microbiol. 2006 nov;4(11):811-25.)。而如果要对这些分泌到宿主细胞内的蛋白进行鉴定则面临诸多挑战,其中之一便是如何从宿主细胞的高丰度蛋白质中鉴定到细菌蛋白。因此就需要可以对宿主细胞内的细菌新产生和新分泌的蛋白进行富集和鉴定的方法。
3.传统的遗传学筛选候选蛋白的策略费时费力且应用范围局限,所以直接针对新生蛋白标记与富集的方法是更能快速识别病原菌效应蛋白的方法。david a. tirrell课题组发明了一种基于非天然氨基酸标记的生物正交方法(boncat),将叠氮基团引入蛋白质中,作为新合成的细胞蛋白质的富集策略(dieterich dc, lee jj, link aj, graumann j, tirrell da, schuman em. labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. nat protoc. 2007;2(3):532-40.),具体的其是通过改造甲硫氨酸的trna合成酶,使其能够识别甲硫氨酸的非天然氨基酸类似物,从而在蛋白合成过程中添加上非天然氨基酸标记,可以阐明蛋白质组的时空动力学变化特征。但是该方法的不足之处在于,其是基于所有蛋白都至少含有一个甲硫氨酸的前提下,也即蛋白起始密码子位(atg)编码的甲硫氨酸(met),但是该位的甲硫氨酸在大多数情况下会被切掉(giglione c, vallon o, meinnel t. control of protein life-span by n-terminal methionine excision. embo j. 2003 jan 2;22(1):13-23. giglione c, boularot a, meinnel t. protein n-terminal methionine excision. cell mol life sci. 2004 jun;61(12):1455-74),从而降低了非天然氨基酸的标记效率。
4.howard c.hang课题组对该方法进行改进,将甲硫氨酸trna合成酶,换成了苯丙氨酸trna合成酶(grammel m, dossa pd, taylor-salmon e, hang hc. cell-selective labeling of bacterial proteomes with an orthogonal phenylalanine amino acid reporter. chem commun (camb). 2012 feb 1;48(10):1473-4)。在除了首位的met外,其他位置苯丙氨酸(phe)的出现几率为3.9%,大于met的2.8%,因此,苯丙氨酸类似物可以相应的标记更多细菌蛋白。但howard c.hang课题组在标记中使用的非天然氨基酸为pep(para-ethynylphenylalanine),该非天然氨基酸会与宿主细胞发生非特异性结合,引入非必要的
背景,且其方法中采用的质粒表达突变型苯丙氨酸trna合成酶的方式,存在表达不稳定,且在宿主细胞内表达易丢失,导致后期蛋白质标记效率降低的问题。
技术实现要素:
5.本发明的目的是提供一种全新的、稳定的细菌新生蛋白标记的方法,以解决现有方法标记不稳定、易丢失的问题。
6.为解决上述技术问题,本发明提供一种稳定标记细菌新生蛋白的方法,通过将细菌的氨酰trna合成酶进行突变,重组,表达,结合非天然氨基酸嵌入,实现对任一空间与时段的细菌新生蛋白的标记。
7.其中,所述非天然氨基酸为苯丙氨酸类似物,优选4-叠氮-苯丙氨酸(azf)。
8.其中,氨酰trna合成酶为大肠杆菌e.coli的苯丙氨酸trna合成酶(phes)。
9.其中,突变位点为phes酶的第294位氨基酸,由丙氨酸(ala)经过点突变为甘氨酸(gly),获得突变后的苯丙氨酸trna合成酶。
10.其中,重组是为将突变后的苯丙氨酸trna合成酶通过λ-red技术嵌入到细菌基因组的attb位点上。
11.其中,非天然氨基酸嵌入通过在细菌培养或生长过程中向培养基中加入一定浓度的azf来实现。
12.所述稳定标记细菌新生蛋白的方法具体包括:第一步,构建可以识别非天然氨基酸的突变型苯丙氨酸trna合成酶phers*基因片段;第二步,将第一步获得的基因片段重组到细菌的基因组上,实现突变型苯丙氨酸trna合成酶的稳定表达;第三步,测试突变型苯丙氨酸trna合成酶不同环境及时长下对非天然氨基酸的嵌入效率。
13.所述第一步进一步包括:第1.1步,从大肠杆菌e.coli菌株基因组中获得完整的phes基因;第1.2步,将第一步中获得的phes基因插入到puc19克隆载体上,以该质粒为模板,通过pcr方法对phes基因进行定点突变,获得phers*基因。
14.所述第二步进一步包括:第2.1步,同样通过设计引物进行pcr扩增的方式,在phers*基因两侧连接细菌attb位点的两个同源臂;第2.2步,先将表达同源重组酶的质粒转入细菌,制备带有该质粒的感受态,其后将第2.1步中的带有attb同源臂的phers*片段电转进入细菌感受态中, 通过同源重组将phers*整合到细菌基因组的attb位点,获得表达phers*的细菌;第三步进一步具体为将第二步中的稳定表达phers*的细菌于m9或rdm培养基中培养或将该细菌侵入宿主细胞后,在培养基中加入不同比例的非天然氨基酸,也即苯丙氨酸类似物4-叠氮-苯丙氨酸(azf),培养一定时间后,进行细菌蛋白提取,通过cu
+
离子依赖的click反应检测苯丙氨酸类似物对细菌新生蛋白的标记效果。
15.本发明的有益效果。
16.本发明提供了一种新的稳定的细菌新生蛋白标记的方法,可以解决现有方法不能对任意时空中(诸如哺乳动物细胞内)的细菌蛋白标记或标记不完全的问题。
附图说明
17.图1:phers*以质粒形式在沙门氏菌内表达情况检测(his-tag);图2:phers*以质粒形式在沙门氏菌内表达后,嵌入非天然氨基酸azf效果检测;图3:phes及phers*以单拷贝在沙门氏菌基因组上的attb位点重组后的表达检测;图4:可在基因组上稳定表达phers*的细菌嵌入非天然氨基酸azf后的效果检测;图5:细胞内沙门氏菌新生蛋白标记效果检测。
具体实施方式
18.鉴于以往的方法对细菌新生蛋白标记不稳定,易造成前期可实现新生蛋白标记,但后期标记缺失,或者无法实现对某些特殊环境下,诸如宿主细胞内的细菌新生蛋白稳定标记的效果,本发明提出了一种细菌新生蛋白的稳定标记方法,标记物为苯丙氨酸类似物,优选4-叠氮-苯丙氨酸(4-azido-l-phenylalanine又称p-azido-phenylalanine,azf),用于提高细菌新生蛋白标记效率,并实现对宿主细胞内任意时段的细菌新生蛋白的稳定标记。
19.该方法包括:第一步,构建可以识别非天然氨基酸的突变型苯丙氨酸trna合成酶基因片段(以下用phers*来代表该基因);第二步,将第一步获得的基因片段重组到细菌的基因组上,实现phers*的稳定表达;第三步,测试表达phers* 的细菌在不同环境、不同azf浓度及不同的标记时间下对非天然氨基酸的嵌入效率。
20.本发明通过大量实验验证得知,在摇瓶培养的条件下,被标记的细菌蛋白总量随着标记时长延长或非天然氨基酸浓度升高而增加,通过实验及分析,发现相对较合适的非天然氨基酸的浓度为50μm~150μm,进一步优选100μm,标记时间为1~3小时,进一步优选 2小时。而在细菌侵入宿主细胞内部之后,非天然氨基酸的标记浓度相应的有所提高,达到1mm,标记时间也提高到3小时。
21.为了实现对细菌新生蛋白的稳定标记,首先要获得稳定的非天然氨基酸嵌入系统,该系统主要包括一个可以在细菌中稳定表达的突变型苯丙氨酸trna合成酶(phers*)和非天然氨基酸的充足供应。前者需要通过基因工程方法将phers*插入到细菌基因组上的attb位点上,该位点是细菌染色体上的附着点,常被用作位点专一性重组。而后在细菌生长或在宿主细胞内定殖的过程中,向培养基中添加足量的非天然氨基酸,这些非天然氨基酸与天然的苯丙氨酸形成竞争,被phers*识别并添加到蛋白合成的肽段上,从而完成了新生蛋白的非天然氨基酸的稳定标记。
22.所述第一步进一步包括:第1.1步,从大肠杆菌e.coli菌株基因组中获得完整的phes基因;第1.2步,将第一步中获得的phes基因插入到puc19克隆载体上,获得质粒,以该质
粒为模板,通过pcr方法对phes基因进行定点突变,获得phers*基因,并通过融合pcr的方式在其两侧添加表达与筛选元件。
23.所述第1.1步进一步具体为:先通过pcr方法,以大肠杆菌e.coli 的mg1655菌株基因组为模板,设计引物扩增其苯丙氨酸trna合成酶基因(以下用phes表示该基因)的全基因片段,并在引物两端引入两个不同的酶切位点。
24.该步中扩增phes全基因片段的引物序列(下划线部分为酶切位点序列):上游引物:5
’‑ꢀ
gaattccactaaagaggagaaaaaaaccatgtcacatctcgcagaa-3’;下游引物: 5
’‑
ggatcccactgaatttcataatctattcctgccttatttaaactgtttgaggaaacgc-3’。
25.苯丙氨酸trna合成酶phes基因的核苷酸序列为:atgtcacatc tcgcagaact ggttgccagt gcgaaggcgg ccattagcca ggcgtcagat gttgccgcgt tagataatgt gcgcgtcgaa tatttgggta aaaaagggca cttaaccctt cagatgacga ccctgcgtga gctgccgcca gaagagcgtc cggcagctgg tgcggttatc aacgaagcga aagagcaggt tcagcaggcg ctgaatgcgc gtaaagcgga actggaaagc gctgcactga atgcgcgtct ggcggcggaa acgattgatg tctctctgcc aggtcgtcgc attgaaaacg gcggtctgca tccggttacc cgtaccatcg accgtatcga aagtttcttc ggtgagcttg gctttaccgt ggcaaccggg ccggaaatcg aagacgatta tcataacttc gatgctctga acattcctgg tcaccacccg gcgcgcgctg accacgacac tttctggttt gacactaccc gcctgctgcg tacccagacc tctggcgtac agatccgcac catgaaagcc cagcagccac cgattcgtat catcgcgcct ggccgtgttt atcgtaacga ctacgaccag actcacacgc cgatgttcca tcagatggaa ggtctgattg ttgataccaa catcagcttt accaacctga aaggcacgct gcacgacttc ctgcgtaact tctttgagga agatttgcag attcgcttcc gtccttccta cttcccgttt accgaacctt ctgcagaagt ggacgtcatg ggtaaaaacg gtaaatggct ggaagtgctg ggctgcggga tggtgcatcc gaacgtgttg cgtaacgttg gcatcgaccc ggaagtttac tctggtttcg ccttcgggat ggggatggag cgtctgacta tgttgcgtta cggcgtcacc gacctgcgtt cattcttcga aaacgatctg cgtttcctca aacagtttaa ataa所述第1.2步进一步具体包括:第1.2.1步,通过酶切连接的方式将该片段连接到puc19克隆载体上;第1.2.2步,以该质粒为模板通过pcr方法,或者利用商业的定点突变试剂盒等手段构建该基因的第294位氨基酸的单碱基定点突变,将该位的氨基酸由gcc(ala)突变为ggc(gly),获得突变后的苯丙氨酸trna合成酶基因(phers*);所述pcr方法构建点突变所需引物如下(下划线位置为点突变碱基):上游引物:5
’‑
gtttactctggtttcggcttcgggatggggatg-3’下游引物:5
’‑
catccccatcccgaagccgaaaccagagtaaac-3’第1.2.3步,对上述突变基因进行测序,确保得到正确的突变序列;第1.2.4步,通过融合pcr的方法在第1.2.3步骤中获得的突变基因基础上,在其前端加上用于表达phers*基因的启动子,rbs区和表达phers*基因的检测标签his-tag,并在其尾端加上用于筛选的荧光序列及抗性基因,及其相应的启动子、rbs区及终止子序列。
26.突变后的苯丙氨酸trna合成酶基因(phers*)核苷酸序列如下(下划线部分为突变的氨基酸密码子,由原来的gcc突变为ggc):
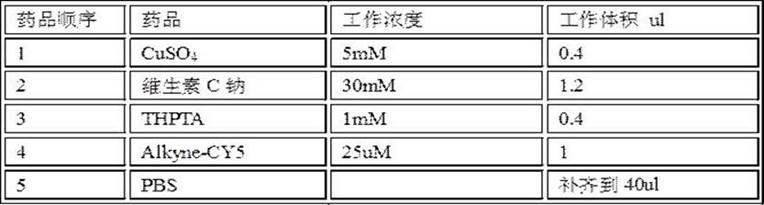
所述第2.2步所述同源重组具体包括:第2.2.1步,将psim6或psim5质粒先电转入细菌中,该质粒可以表达同源重组酶,而需注意的是,为防止质粒丢失,带有该质粒的细菌应该在25℃~30℃培养,优选30℃;第2.2.2步,挑取携带有psim6或psim5质粒的细菌单克隆,接种于lb培养基中,同时加入100ug/ml氨苄青霉素(amp)或25ug/ml氯霉素作为抗性筛选( psim6 带amp抗性而psim5带氯霉素抗性,需依据不同质粒使用对应的抗生素),于25℃~30℃,优选30℃,150-200rpm摇床,过夜培养;第2.2.3步,将2.2.2中的菌液1:30转接到100ml含有100ug/ml氨苄青霉素或25ug/ml氯霉素的新鲜lb中,于30℃,150-200rpm摇床,培养至od600=0.5左右;第2.2.4步,将2.2.3中的菌液继续以1:30转接到100ml含有100ug/ml氨苄青霉素或25ug/ml氯霉素的新鲜lb中,于30℃,150-200rpm摇床,培养至od600=0.5左右,制备带有psim6或psim5质粒细菌电转感受态;第2.2.5步,将第2.1步中获得的带有attb同源臂的phers*基因及其表达与筛选元件的片段,通过电转的方式,转入2.2.4中的细菌感受态中,加入1ml预热的lb培养基后于30℃,150-200rpm孵育1小时后,取100ul涂板(平板上的固体lb 培养基中带有卡那霉素加氨苄青霉素(或卡那霉素加氯霉素)两种抗生素),于30℃细菌培养箱中培养过夜;第2.2.6步,挑取2.5中lb平板上的单克隆,接种于新鲜的、仅含有50ug/ml卡那霉素的lb培养基中,于37℃,150-200rpm摇床培养过夜,该步骤是为了,主动让细菌丢失psim6或psim5质粒,同时,筛选已经将带有卡那霉素抗性的phers*基因片段整合到细菌基因组上的菌株;第2.2.7步,设计验证引物,将菌液通过pcr进行验证,同时将pcr产物送测序验证,结果正确后,获得了在细菌基因组attb位点插入了phers*基因片段的菌株。
30.所述第2.2.7步中验证引物如下:phers*基因及其表达与筛选元件的片段在沙门氏菌stm14028s菌株基因组attb位点重组后的验证引物:上游引物:5
’‑
agtgttgagc atcgaaattc-3’下游引物:5
’‑ꢀ
cgtattgatg tcgatgaagg-3’phers*基因及其表达与筛选元件的片段在大肠杆菌mg1655菌株基因组attb位点重组后验证引物:上游引物:5
’‑ꢀ
gggaaaccga taatgtattc ac-3’下游引物:5
’‑ꢀ
gtttccggcc tgaaaaggaa ct-3’所述第三步进一步具体为,将上一步中的带有phers*酶的细菌于m9或rdm培养基培养或将细菌侵入宿主细胞后,在培养基中加入不同比例的非天然氨基酸azf,培养一定时间后,进行细菌蛋白提取,通过cu
+
离子依赖的click反应检测azf对细菌新生蛋白的标记效果。
31.所述第三步进一步具体为:第3.1步,挑取带有phers* 的细菌单克隆,接种于带有50ug/ml卡那霉素的lb培养基中,于37℃,150-200rpm摇床培养过夜;第3.2步,将3.1中的菌液以1:30转接于新鲜的含有50ug/ml卡那霉素的lb培养基
中,于37℃,150-200rpm摇床培养至平台期;第3.3步,通过离心收集细菌,重悬于m9培养基中,于37℃,150-200rpm培养15分钟,再次离心收集细菌;第3.4步,将上一步中的菌体以对应最初体积的m9重悬,并加入非天然氨基酸azf,此处设置不同浓度,例如1μm、10μm、100μm和1mm,并于37℃,150-200rpm培养条件下,标记不同时间,例如0.5h、1h、1.5h和2h等;第3.5步,将上一步中的细菌再次离心收集菌体,用pbs洗涤一次,而后依据菌体大小,加入适量的2%sds(sds溶解于ddh2o中)重悬,并在其中加入1x蛋白酶抑制剂充分混匀;第3.6步,进行蛋白提取,超声破碎的方法,破碎菌体和dna并释放细菌蛋白,超声设置为:60%power,3min/次,3s on/3s off;每个样品超声一次;第3.7步,将超声后的蛋白以最大转速离心10分钟后,吸取上清进行bca检测浓度;第3.8步,蛋白以不超过1μg/μl的浓度进行叠氮与炔基的环加成反应,又称click反应,用以对非天然氨基酸标记的蛋白进行检测。
32.所述第3.5步中,蛋白抑制剂可以为complete
™
, edta-free protease inhibitor cocktail。
33.以下采用实施例和附图来详细说明本发明的实施方式,借此对本发明如何应用技术手段来解决技术问题,并达成技术效果的实现过程能充分理解并据以实施。
34.实施例1获得完整的突变型苯丙氨酸trna合成酶基因,及其表达与筛选元件a、首先通过pcr方法,以大肠杆菌e.coli m1655菌株基因组为模板,扩增其苯丙氨酸trna合成酶基因(phes)的cds编码区片段,并在引物两端引入两个不同的酶切位点;b、通过酶切连接的方式将该片段连接到克隆载体上。
35.c、以b中的质粒为模板通过pcr的方法构建该基因的第294位氨基酸的单碱基定点突变,将该位的氨基酸由gcc(ala)突变为ggc(gly),获得突变后的苯丙氨酸trna合成酶基因(phers*);并用同样的手段在其尾端加上mcherry荧光基团序列以及卡那霉素抗性基因及其启动子及终止子序列,用同源重组后的筛选。
36.d、设计引物,使得上下游引物5’端各带有50bp的细菌attb位点的同源臂片段,通过pcr扩增,最终得到带有attb同源臂的phers*。
37.e、将上述片段送去测序,正确后进行下一步实验。
38.构建稳定表达phers*的沙门氏菌菌株首先将psim6质粒先电转入鼠伤寒沙门氏菌野生型菌株14028s菌株(stm14028s)中,注意所有涉及到该菌株的操作均应该在bsl-2实验室进行。该质粒可以表达同源重组酶,而需注意的是,为防止质粒丢失,带有该质粒的细菌应该在30℃培养;挑取携带有psim6质粒的stm14028s菌株单克隆,接种于lb培养基中,同时加入氨苄青霉素(amp)作为抗性筛选(psim6带amp抗性),于30℃,220rpm摇床,过夜培养;将过夜培养的菌液1:30转接到10ml含有100ug/ml氨苄青霉素的新鲜lb中,于30℃,220rpm rpm摇床,培养至od600=0.5左右;将该菌液继续以1:30转接到100ml含有100ug/ml氨苄青霉素的新鲜lb中,于30℃,150-200rpm摇床,培养至od600=0.5左右,于4℃,4000rpm离心收集菌体,并用同样4℃预冷的ddh2o洗两遍菌体后,制备成带有psim6质粒stm14028s菌株电转感受态;
其次将带有stm14028s菌株attb同源臂的phers*基因片段,通过电转的方式,转入stm14028s菌株感受态中,加入1ml预热的lb培养基后于30℃,150-200rpm孵育1小时后,取100ul涂板(平板上的固体lb 培养基中带有卡那霉素和氨苄青霉素两种抗生素),于30℃细菌培养箱中培养过夜;挑取lb平板上的单克隆,接种于新鲜的、仅含有50ug/ml卡那霉素的lb培养基中,于37℃,150-200rpm摇床培养过夜,该步骤是为了,主动让细菌丢失psim6质粒,同时,筛选已经将带有卡那霉素抗性的phers*基因片段整合到细菌基因组上的菌株;此时可依据stm14028s菌株的attb位点设计一对检测引物,同时利用未插入phers*基因片段的stm14028s菌株做对照,通过菌液pcr进行验证,也为了避免其他菌株的污染,可同时依据stm14028s菌株的16s设计引物,进行菌株检测,并将pcr产物送测序验证,结果正确后,则获得了可以稳定表达phers*的沙门氏菌。
39.细胞外(摇瓶培养基中)沙门氏菌新生蛋白的标记挑取稳定表达phers*的stm14028s菌株单克隆,接种于带有50ug/ml卡那霉素的lb培养基中,于37℃,220rpm摇床培养过夜;菌液以1:30转接于新鲜的含有50ug/ml卡那霉素的lb培养基中,于37℃,220rpm摇床培养至平台期;通过离心收集细菌,重悬于m9培养基中,于37℃,220rpm培养15分钟,再次离心收集细菌;将菌体以对应最初体积的m9重悬,并加入非天然氨基酸azf,此处设置不同浓度,例如1μm、10μm、100μm和1mm,并于37℃,220rpm培养条件下,标记不同时间,例如15min、30min、45min、60min、90min和120min等;将细菌再次离心收集菌体,用pbs洗一遍,而后依据菌体大小,加入适量的2%sds(sds溶解于ddh2o中)重悬,并在其中加入蛋白酶抑制剂充分混匀;进行蛋白提取,此处通过超声破碎的方法,破碎菌体和dna并释放细菌蛋白,超声设置为:60%power,3min/次,3s on/3s off,超声1次。将超声后的蛋白以最大转速离心10分钟后,吸取上清进行bca检测浓度。蛋白以不超过1μg/μl的浓度进行click反应(反应体系见表2),用以对非天然氨基酸标记的蛋白进行检测。click反应后的蛋白可直接跑sds-page电泳,对被标记蛋白进行胶上的荧光检测。其具体反应体系,见下表2。
40.对于细胞内沙门氏菌新生蛋白标记提前24h将1x106个小鼠巨噬细胞raw264.7接种于铺了盖玻片的六孔板中,加入2ml含有10%fbs和1%青链霉素双抗的dmem培养基,于37℃ 5%的co2培养箱中培养过夜。
41.挑取稳定表达phers*的stm14028s菌株单克隆,接种于带有50ug/ml卡那霉素的lb液体培养基中,于37℃,220rpm摇床培养过夜;菌液稀释后测其od600,沙门氏菌od600=1时,其对应的细菌个数为5x10^8个/ml,pbs洗一遍,后重悬于含有10%fbs的dmem培养基中;以moi=100(moi为感染复数,即细菌与raw264.7细胞个数比为100:1)的比例侵染小鼠巨噬细胞raw264.7,注意此时培养基不添加抗生素,于37℃,5%的co2培养箱中培养30min,而后弃去培养基,并用pbs洗两遍,加入含有100μg/ml庆大霉素的10%fbs的dmem培养基,于37℃,5%的co2培养箱中培养1h,后换成含有16.5μg/ml庆大霉素的10%fbs的dmem培养基,此时开始计时,孵育不同时间,例如计划在孵育4h,8h,12h,16h,20h后取样,那么就需要于取样时间点的提前3h加入1mm azf进行标记,也就是与1h,5h,9h,12h和16h加入azf进行非天然氨基酸标记。
42.具体样品收集与处理是,在特定时间点,将长满细胞的盖玻片(我们称之为细胞爬片)从培养基中取出,并浸于pbs中洗一次,5min/次;而后对细胞进行固定,即将细胞爬片浸
入含有4%pfa(多聚甲醛)的pbs中,室温下孵育10min,使细胞固定在盖玻片上不易脱落,其后的click反应均在细胞爬片上进行。具体为,固定后的细胞爬片浸于含有100mm甘氨酸的pbs中,于室温下孵育5min,此步骤为了淬灭上一步的pfa的作用;而后pbs洗三次,每次5min;之后将细胞爬片于含有0.1%的triton x-100 的pbs中室温下孵育10min,此步骤是为了在细胞膜上打孔,如果后续实验中需要用到抗体的话,那么此步骤可以使抗体能顺利进入细胞内;然后pbs洗3次,每次5min;并依据表1中的click反应配置反应体系,将40ul反应液滴加到平铺的封口膜上,将爬片倒扣于液滴上,使得细胞充分接触液体,不要有气泡,并避光反应1h;pbs洗3次,每次5min;之后可依据实验需要孵育其他抗体或染料(本实施例中不需要用到其他抗体,故可省略此步骤);最后封片,在封片剂中加入万分之一浓度的dapi,混匀后,取12μl封片剂滴加于载玻片上,倒扣上细胞爬片,于4℃过夜封片,及保存,以便后续用于荧光显微镜或共聚焦显微镜观察。
43.上面所述的对细胞内、外的细菌新生蛋白检测方法如下,表1为用于荧光显微镜观察的,爬片上的细胞蛋白click反应体系。
44.表1表2为用于蛋白sds-page检测的click反应体系,标记有azf的蛋白样品在进行click反应前,需95℃加热5min。
45.表2注意事项:a,药品添加顺序严格遵循从上至下的顺序,将维生素c钠加入到硫酸铜后,其颜色立即呈现深棕色,而后在10秒内转为黄色,待其颜色变黄后加入thpta,因其螯合作用,药品颜色会变为透明色,此时再按顺序加入后面所列的药品,进行click反应;b,反应体系可随需要进行调整,体系中蛋白浓度不超过1ug/ul(因铜离子会使得过高的蛋白浓度发生沉淀,进而导致蛋白样品损失);c,样品充分混匀,室温下避光,1h;d,最大转速离心5min,将上清转移至新的离心管中,弃去沉淀;
e,上清样品用于跑蛋白sds-page(10%)胶,并进行蛋白的荧光检测。
46.图3中显示,通过westernblot验证了phers*在插入到细菌基因组attb位点后,是呈现稳定表达的,即使是来源于不同克隆的菌株,其表达phers*的结果也一样。1为不表达任何形式的phers*的野生型沙门氏菌对照组,2-3为attb位点插入原有phes基因后,不同菌株内phes的表达检测,4-5为突变型phers*基因插入后,不同菌株内phers*的表达检测,图中显示,不同菌株在基因组上的phers*表达相对稳定,不会随着抗生素抗性的消失而出现不表达的情况。
47.图4中显示,我们检测图3中稳定表达phers*的沙门氏菌对非天然氨基酸的嵌入情况发现,即使是不同的菌株,在相同的标记情况下,其对细菌新生蛋白的标记模式是一样的,这也进一步说明了,基因组插入phers*的形式更有利于稳定嵌入非天然氨基酸,进而标记细菌新生蛋白。其中,1-2为野生型沙门氏菌和仅表达phes的对照菌株,结果显示其不能有效利用非天然氨基酸标记细菌蛋白,3-4是在基因组上表达phers*的两株不同菌株,对azf的嵌入效果,3、4为不同菌株,但其在相同条件下对非天然氨基酸的嵌入效果一致,说明了基因组表达phers*的稳定性。
48.图5中,我们将图3中的表达phers*沙门氏菌侵染宿主细胞,待细菌进入细胞后,对细胞内的沙门氏菌进行不同时段的非天然氨基酸标记,结果显示,phers*可以实现对不同时段细胞内细菌新生蛋白的非天然氨基酸标记,而且phers*不会因抗生素的消失而不表达。图中从上到下分别为沙门氏菌感染小鼠巨噬细胞后不同时间,其新生蛋白的标记效果检测。第一列为dapi染色的细胞核,第二列为带红色荧光的沙门氏菌菌体,第三列为非天然氨基酸标记的沙门氏菌的新生蛋白,第4列为前3张图的叠加图。图中可见,即使细菌侵入到宿主细胞内,已经不存在抗生素抗性压力,只要细菌仍在生长,那么细菌基因组上稳定表达的phers*都可以利用培养基中的azf,实现对细菌新生蛋白的标记。
49.实施例2以实施例1中的方法构建可以稳定标记新生蛋白的大肠杆菌菌株。具体如下:获得完整的突变型苯丙氨酸trna合成酶基因,及其表达与筛选元件a、首先通过pcr方法,以大肠杆菌e.coli mg1655菌株基因组为模板,扩增其苯丙氨酸trna合成酶基因(phes)的cds编码区片段,并在引物两端引入两个不同的酶切位点;b、通过酶切连接的方式将该片段连接到克隆载体上。
50.c、以b中构建的质粒为模板,通过pcr的方法构建该基因的第294位氨基酸的单碱基定点突变,将该位的氨基酸由gcc(ala)突变为ggc(gly),获得突变后的苯丙氨酸trna合成酶基因(phers*);并用同样的手段在其尾端加上mcherry荧光基团序列以及卡那霉素抗性基因及其启动子及终止子序列,用同源重组后的筛选。
51.d、设计引物,使得上下游引物5’端各带有50bp的细菌attb位点的同源臂片段,通过pcr扩增,最终得到带有attb同源臂的phers*。
52.e、将上述片段送去测序,正确后进行下一步实验。
53.构建稳定表达phers*的大肠杆菌菌株首先将psim5质粒先电转入大肠杆菌e.coli m1655菌株(mg1655)中。该质粒可以表达同源重组酶,而需注意的是,为防止质粒丢失,带有该质粒的细菌应该在30℃培养;挑取携带有psim5质粒的mg1655菌株单克隆,接种于lb培养基中,同时加入氯霉素(cmr)作为
抗性筛选(psim5带氯霉素抗性),于30℃,220rpm摇床,过夜培养;将过夜培养的菌液1:30转接到10ml含有25ug/ml氯霉素的新鲜lb中,于30℃,220rpm rpm摇床,培养至od600=0.5左右;将该菌液继续以1:30转接到100ml含有25ug/ml氯霉素的新鲜lb中,于30℃,150-200rpm摇床,培养至od600=0.5左右,于4℃,4000rpm离心收集菌体,并用同样4℃预冷的ddh2o洗两遍菌体后,制备成带有psim5质粒mg1655菌株电转感受态;其次将带有mg1655菌株attb同源臂的phers*基因片段,通过电转的方式,转入mg1655菌株感受态中,加入1ml预热的lb培养基后于30℃,150-200rpm孵育1小时后,取100ul涂板(平板上的固体lb 培养基中带有卡那霉素和氯霉素两种抗生素),于30℃细菌培养箱中培养过夜;挑取lb平板上的单克隆,接种于新鲜的、仅含有50ug/ml卡那霉素的lb培养基中,于37℃,150-200rpm摇床培养过夜,该步骤是为了,主动让细菌丢失psim5质粒,同时,筛选已经将带有卡那霉素抗性的phers*基因片段整合到细菌基因组上的菌株;此时可依据mg1655菌株的attb位点设计一对检测引物,同时利用未插入phers*基因片段的mg1655菌株做对照,通过菌液pcr进行验证,也为了避免其他菌株的污染,可同时依据mg1655菌株的16s设计引物,进行菌株检测,并将pcr产物送测序验证,结果正确后,则获得了可以稳定表达phers*的大肠杆菌菌株。细胞外(摇瓶培养基中)大肠杆菌新生蛋白的标记挑取稳定表达phers*的mg1655菌株单克隆,接种于带有50ug/ml卡那霉素的lb培养基中,于37℃,220rpm摇床培养过夜;菌液以1:30转接于新鲜的含有50ug/ml卡那霉素的lb培养基中,于37℃,220rpm摇床培养至平台期;通过离心收集细菌,重悬于m9培养基中,于37℃,220rpm培养15分钟,再次离心收集细菌;将菌体以对应最初体积的m9重悬,并加入100μm非天然氨基酸azf,并于37℃,220rpm培养条件下,标记120min;将细菌再次离心收集菌体,用pbs洗一遍,而后依据菌体大小,加入适量的2%sds(sds溶解于ddh2o中)重悬,并在其中加入1x蛋白酶抑制剂充分混匀;进行蛋白提取,此处通过超声破碎的方法,破碎菌体和dna并释放细菌蛋白,超声设置为:60% 功率,3min/次,3s on/3s off,超声1次。将超声后的蛋白以最大转速离心10分钟后,吸取上清进行bca检测浓度。蛋白以不超过1μg/μl的浓度进行click反应,用以对非天然氨基酸标记的蛋白进行检测。click反应后的蛋白可直接跑sds-page电泳,进行被标记蛋白的荧光检测。其具体反应体系,见表2。
54.比较例1依据文献(grammel m, dossa pd, taylor-salmon e, hang hc. cell-selective labeling of bacterial proteomes with an orthogonal phenylalanine amino acid reporter. chem commun (camb). 2012 feb 1;48(10):1473-4)所述,采用pcr方法,以大肠杆菌e.coli k12菌株基因组为模板,通过引物扩增其苯丙氨酸trna合成酶基因(phes)的cds编码区片段。
55.引物核苷酸序列为:gagctccatgtcacatctcgcagaactgg,ggtacccgcatcgctatcaatcgcc将该片段亚克隆到pjet1.2克隆载体上,通过位点特异性突变试剂盒进行定点突变,构建该基因的第294位氨基酸的单碱基定点突变,将该位的氨基酸由gcc(ala)突变为ggc(gly),获得突变后的苯丙氨酸trna合成酶基因(phers*),其后通过引入两个不同的酶切位点(saci 和kpni)将该片段插入到pwsk29表达质粒上。
56.最后将该质粒通过电转进鼠伤寒沙门氏菌 salmonella typhimurium ir715 菌株中。通过氨苄青霉素抗性筛选,获得表达phers*的沙门氏菌。
57.挑取单克隆菌落,接种于含有100ug/ml羧苄青霉素的lb液体培养基中,过夜培养,而后1:30转接新鲜的含有100ug/ml羧苄青霉素的lb液体培养基,于37℃培养至平台期。于10,000rpm,3min收集细菌,并重悬于m9培养基中(同样含有100ug/ml羧苄青霉素)。在m9培养基中培养15min后,同样离心收集菌体,以相应体积重悬于m9培养基中,并向其中加入一定比例的非天然氨基酸,标记1h后,重新收集菌体,并用pbs洗一遍。
58.菌体经过同实例1中相同的裂解和超声后,提取蛋白,并进行蛋白表达检测及click反应检测。
59.图1中我们利用westernblot对质粒表达的phers*进行了检测,发现不同菌株表达phers*是存在显著差异的,这就影响了其后续对非天然氨基酸的嵌入效果。其中,1:不表达phers*的野生型菌株对照,2-5为表达同一质粒的不同菌株,不同菌株中phers*的表达存在差异,说明质粒表达的phers*存在表达不稳定的问题。p-a294g为phers*的表达质粒。
60.图2中,我们对图1中的菌嵌入非天然氨基酸标记细菌新生蛋白的能力进行检测,发现不同菌株因表达的phers*量不同,其嵌入非天然氨基酸的效果也会出现显著差异,进而直接影响对细菌蛋白的标记效果。图中所示,1-2分别为野生型和空载质粒对照组,3-4为表达同一质粒的不同菌株嵌入非天然氨基酸azf的效果,因不同菌株中phers*质粒的表达尚且存在差异,那么其对应的嵌入非天然氨基酸的能力也会相应的出现差异,且质粒在抗生素抗性筛选压力消失时易丢失,那么则不能对细菌的新生蛋白进行有效标记。
61.通过图1与图3的比较可以看出,以质粒形式表达的phers*,依据不同质粒的特性,诸如质粒在细菌中的拷贝数,启动子和rbs区强度等,存在表达不稳定的情况,也即不同菌株phers*的表达量会有所差异;而且质粒在细菌生长过程中容易丢失,尤其是如果细菌侵入宿主细胞后,因其失去了抗生素抗性筛选的压力,质粒会在其后的数小时内陆续丢失;这意味着,菌株嵌入非天然氨基酸的能力会下降或不稳定,诸如图2中所示,菌株3和4号,虽然表达的是同一个phers*质粒,但相同条件下对非天然氨基酸的嵌入效果却不同,这就很难保证对不同生长条件下的细菌的新生蛋白的稳定标记。相反,本技术中,将phers*插入到细菌的基因组上,首先保证了其拷贝数的稳定,也即表达稳定,正如图3中所示,不同菌株的phers*表达相同,而且其表达不会随着细菌的生长环境变化发生波动,即使在抗生素筛选压力不存在时,仍然可以表达。图4显示其结合非天然氨基酸情况,可见,即使是不同菌株,但在稳定表达phers*的前提下,是可以对非天然氨基酸实现稳定嵌入的。
62.所有上述的首要实施这一知识产权,并没有设定限制其他形式的实施这种新产品和/或新方法。本领域技术人员将利用这一重要信息,上述内容修改,以实现类似的执行情况。但是,所有修改或改造基于本发明新产品属于保留的权利。
63.以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1