检测PRCC/TFE3融合基因的荧光原位杂交探针组及其制备方法和应用与流程
检测prcc/tfe3融合基因的荧光原位杂交探针组及其制备方法和应用
技术领域
1.本发明涉及基因检测技术领域,具体涉及一种检测prcc/tfe3基因的荧光原位杂交探针组及其制备方法和应用。
背景技术:
2.xp11.2易位性肾细胞癌(trcc)是一种罕见的肾细胞癌(rcc)亚型,是tfe3转录因子基因融合的结果,在世界卫生组织(who)泌尿系统肿瘤分类中被纳入mit家族trcc。在肾癌中,xp11.2 trcc占儿童肾肿瘤的20%至75%,约占成人肾细胞癌的1.5%。xp11.2 trcc中最常见的基因融合是xp11.2上的tfe3基因与1q21位点的prcc基因发生融合,tfe3基因与17q25位点的aspl基因发生融合,这些基因融合来自于t(x;1)(p11.2;q12)和t(x;17)(p11.2;q25.3)易位,其他发生较少的tfe3融合伙伴包括sfpq、nono、cltc和rbm10,它们由t(x;1)(p11.2;p34),inv(x)(p11.2q12),t(x;17)(p11.2;q23)和inv(x)(p11.2p11.23)引起,一些融合伙伴,如parp14,khtfesrp,luc7l3和dvl2,仅在个别患者中被发现。
3.荧光原位杂交实验可以用于评估prcc/tfe3基因融合状态。传统的商业化fish探针主要使用bac克隆作为探针,经过荧光分子标记后进行杂交检测,然而,bac克隆制备的探针存在一些非重复序列,探针特异性不高,需要使用cot1-dna进行非封闭性封闭,且bac克隆制备的探针片段普遍偏大,长度在200~500bp之间,杂交效率偏低,杂交时间长。
技术实现要素:
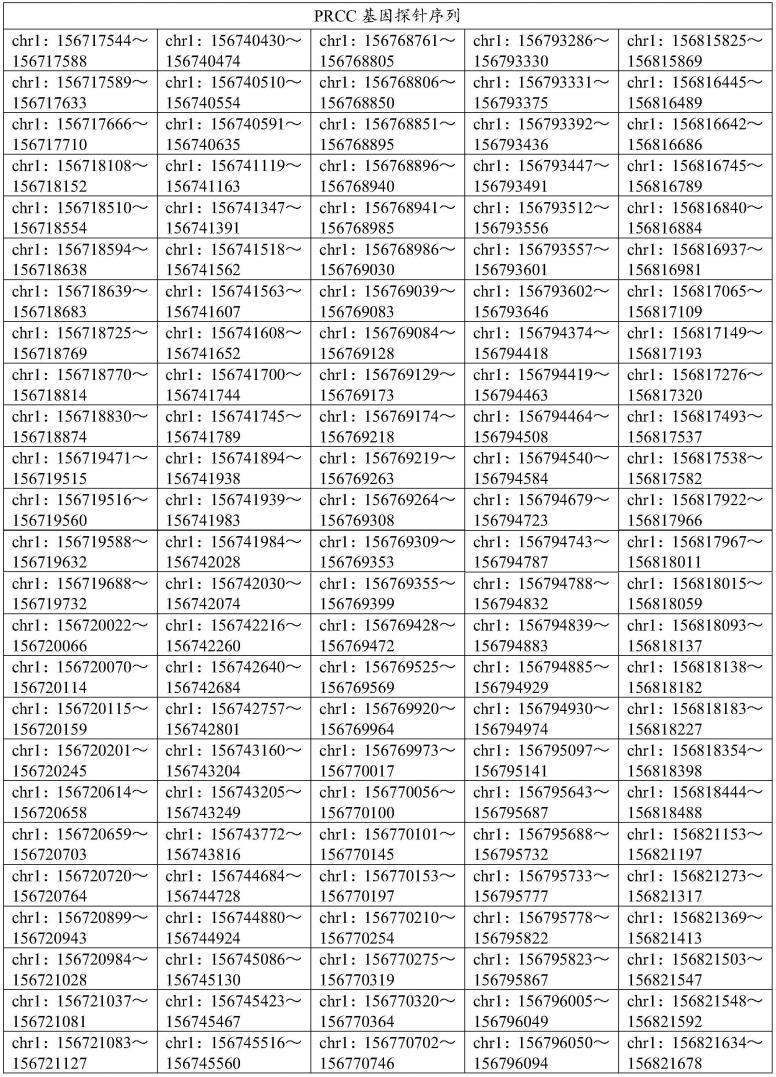
4.针对现有技术所存在的技术问题,本发明的目的在于提供一种检测prcc/tfe3融合基因的荧光原位杂交探针组,该探针组由说明书表1中所示的1533个核苷酸序列制备得到。
5.作为一种优选的实施方式,该探针组由上述的核苷酸序列通过添加标签序列后扩增得到。
6.作为一种优选的实施方式,所述核苷酸序列的5’端标签序列为:taatacgactcactataggg;3’端标签序列为:ccgctgagcaataactagca。
7.作为一种优选的实施方式,pcr扩增体系中含有alexa fluor555-dutp和alexa fluor488-dutp。
8.本发明还提供了上述探针组的制备方法,包括以下步骤:
9.s1,以prcc和tfe3基因序列为依据,在不含重复序列的区域分别设计含45个碱基核苷酸序列,设定探针tm为50℃,gc含量范围为40~60%,舍弃包含aaaa/tttt/cccc/gggg的序列后,得到2001条候选探针,再经过全基因hg38 blast比对分析后,最终得到1533个核苷酸序列,具体参见说明书表1所示;
10.s2,分别在每条核苷酸序列的5’端加上20bp的标签序列、3’端加上20bp的标签序列,得到一系列带有标签序列的特征性引物,其中5’端标签序列为:taatacgactcactataggg,3’端标签序列为:ccgctgagcaataactagca;
fluor488-dutp标记的dutp进行pcr扩增,将荧光标记材料掺入pcr产物中,得到荧光标记的pcr产物;pcr扩增时的引物为:primer-f:taatacgactcactatagg,primer-r:tgctagttattgctcagcgg;
27.pcr扩增体系为:10x buffer 5μl、f+r primer(10um)2μl、洗脱后特征性引物1μl、10mm datp 1μl、10mm dctp 1μl、10mm dgtp 1μl、10mm dttp 0.75μl、1mm alexa fluor555-dutp 2.5μl、taq dna polymerase 0.5μl、ddh20 35.25μl;
28.pcr反应程序为94℃5min、55℃5min;72℃2min、94℃1min、55℃1min,30个循环;72℃5min、4℃保温。
29.s4,对步骤s3扩增后的pcr产物进行纯化,得到荧光原位杂交探针组。
30.表1 1533条探针的序列信息
31.32.33.34.35.36.37.38.39.40.41.[0042][0043]
将上述制备得到的探针组按照下表2的组分混合制备得到杂交缓冲液,并进一步制备成试剂盒。
[0044]
表2 10人份的杂交缓冲液的制备
[0045]
成分10人份终浓度实施例1的探针组合10μl20ng/μl(各探针)去离子甲酰胺45μl45%50%葡聚糖20μl10%20
×
ssc10μl2
×
ssc(0.2m)tris-hcl(ph8.0~8.8)5μl50mm水10μl-[0046]
实施例2正常人外周淋巴细胞滴片实验
[0047]
材料:人外周血淋巴细胞培养基、秋水仙碱、低渗液(质量体积百分比为0.4%的kcl)、固定液(甲醇:乙酸=3:1,体积比)。
[0048]
s1,细胞培养及同步化:取0.4ml肝素抗凝全血于人外周血淋巴细胞培养基中,混匀后于37℃、5%co2恒温培养箱内培养72h,并于终止前4h,向培养基中加入秋水仙素至终浓度(0.1μg/ml)继续培养4h;
[0049]
s2,收集和固定:收集培养基,500g离心5min后,弃上清,再加入0.4%kcl低渗液孵育30min,然后用甲醇-冰醋酸固定液固定细胞,室温静置10min,500g离心沉淀细胞,重复细胞固定步骤一遍,弃上清液后,根据细胞数量加入适当固定液悬浮细胞;
[0050]
s3,制片:吸取7-10μl细胞悬液,自70-80cm高处滴在一张干燥清洁的载玻片上,吹干后,56℃烤片2h;
[0051]
s4,变性杂交:取10μl实施例1中制得的杂交缓冲液滴加到处理好的样本的玻片杂交区域,立即加盖盖玻片,用橡皮胶封边,避免盖玻片与玻片之间产生气泡,杂交仪上95℃共变性2min,42℃杂交30min;
[0052]
s5,杂交后洗涤:用镊子小心撕去盖玻片周围封片胶,避免粘掉或者移动盖玻片;将载玻片放入稀释后的洗涤液1(含0.6m氯化钠和50mm tris-hcl)中浸泡1min,待盖玻片自然脱落,再将载玻片放入洗涤液2(含0.6m氯化钠、50mm tris-hcl和0.3%np-40)于52℃中浸泡5min,最后将载玻片放入37℃预热的去离子水中浸泡10s,暗处自然干燥载玻片;
[0053]
s6,dapi(4',6-二脒基-2-苯基吲哚)复染:向切片正中心滴加细胞核染色液(荧光原位杂交样品处理试剂盒细胞核染色液,购自thermo fisher scientific),盖玻片封片,置于荧光显微镜下,先在低倍物镜(10
×
)下确认细胞区域;转到40
×
物镜下,找到一个细胞分布均匀的位置,再在高倍物镜(100
×
)下观察细胞核的fish结果。
[0054]
检测含有50个中期淋巴细胞的载玻片,结果如下表3所示,可以看出,prcc基因探针的fish信号点都在99个,敏感性达到99%;tfe3基因探针的fish信号点都在99个,敏感性达到99%
[0055]
表3探针的敏感性检测结果
[0056][0057]
实施例3组织切片prcc/tfe3融合基因fish实验
[0058]
s1,石蜡组织切片前处理:将肾组织切片置于烤片机上65℃烤片1-2小时;将组织切片放入二甲苯中室温脱蜡10分钟,重复一次,然后放入100%无水乙醇中室温10分钟,去除残留二甲苯;取出玻片,将其放入100%、90%、70%梯度乙醇中各2分钟,随即浸入去离子水中3分钟,取出切片后用无绒纸巾沿组织周围吸去多余水分;再将其放入通透剂(10mm tris-base、1mm edta、0.2%triton x-100)中90℃煮片20分钟;取出玻片,再将玻片放入去离子水中室温洗涤5分钟;再将玻片放入37℃预热的蛋白酶k工作液中,消化20-30分钟;取出玻片将其放入2
×
ssc中室温洗涤5分钟;取出玻片后,再放入另一缸2
×
ssc中室温洗涤5分钟;再将玻片依次放入70%,90%,100%梯度乙醇室温脱水各3分钟;取出玻片,室温晾干。
[0059]
s2,变性杂交:取10μl实施例1制得的杂交缓冲液滴加到处理好的样本的玻片杂交区域,立即加盖盖玻片,用橡皮胶封边,避免盖玻片与玻片之间产生气泡;杂交仪上95℃共变性2min,42℃杂交30min。
[0060]
s3,杂交后洗涤:用镊子小心撕去盖玻片周围封片胶,避免粘掉或者移动盖玻片;将载玻片放入稀释后的洗涤液1(含0.6m氯化钠和50mm tris-hcl)中浸泡1min,待盖玻片自然脱落,再将载玻片放入洗涤液2(含0.6m氯化钠、50mm tris-hcl和0.3%np-40)于52℃中浸泡5min,最后将载玻片放入37℃预热的去离子水中浸泡10s,暗处自然干燥载玻片。
[0061]
s4,dapi复染:向切片正中心滴加细胞核染色液(荧光原位杂交样品处理试剂盒细胞核染色液),盖玻片封片,置于荧光显微镜下,先在低倍物镜(10
×
)下确认细胞区域;转到40
×
物镜下,找到一个细胞分布均匀的位置,再在高倍物镜(100
×
)下观察细胞核的fish结果。
[0062]
实验结果:结果如图1所示,组织细胞中prcc和tfe3基因探针信号明亮清晰可见,敏感性高。
[0063]
在此有必要指出的是,以上实施例仅限于对本发明的技术方案做进一步的阐述和说明,并不是对本发明的技术方案的进一步的限制,本发明的方法仅为较佳的实施方案,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1