一种嘧啶联苯芳香环化合物的制备方法与流程
1.本技术涉及液晶材料的制备领域,更具体地说,它涉及一种嘧啶联苯芳香环化合物的制备方法。
背景技术:
2.液晶材料是一种介晶相的高分子材料,其分子间作用力比固体弱,微小的外部能量如电场、磁场、热能等即可实现各分子状态间的转变,并引起它的光、电、磁的物理性质发生变化,上述特性使得显示、调光等光学领域获得了迅猛的发展。
3.现阶段采用联苯类和联三苯类作为液晶材料,由于其末端基团为烷烃基联苯化合物,具有五色、化学性能稳定、光化学性能稳定、介电各向异性及粘度和折射率适中的特点,被广泛的应用于lcd、oled领域。
4.本技术仅以应用最为普遍的嘧啶联苯化合物为例,相关技术中嘧啶联苯化合物的制备方法主要是通过酰胺与酰胺之间关环得到嘧啶环,然后通过r1的格式试剂直接与吡啶环上的卤素偶联,反应得到目标产物。
5.上述制备方法虽也能得到目标产物,但产物收率极低,且不易分离,此外其过程繁杂往往需要三到四步反应的同时,对化合物的各方面要求苛刻,制备方法无法适用于所有的嘧啶联苯化合物。
6.综上,本技术特提供一种反应干净、收率高、且产物易于分离的嘧啶联苯芳香环化合物的制备方法。
技术实现要素:
7.为解决上述技术问题,本技术提供一种嘧啶联苯芳香环化合物的制备方法,该制备方法反应干净、收率高的同时,产物易于分离。
8.第一方面,本技术提供一种嘧啶联苯芳香环化合物的制备方法,采用如下的技术方案:一种嘧啶联苯芳香环化合物的制备方法,步骤如下:s1、先将卤代嘧啶与含有r2基团的联苯硼酸进行偶联反应,得主产物;其中r2基团选自
‑ꢀ
ch3(ch2)2、-ch3(ch2)3、-ch3(ch2)4或-ch3(ch2)5;s2、再将s1中所得主产物与含有r1基团的炔类化合物在钯配体和cui的催化下反应,得次级产物;其中r1基团选自-ch3(ch2)4、-ch3(ch2)5、-ch3(ch2)6、-ch3(ch2)7或-oh;s3、最后对s2中所得的次级产物进行氢化反应,即可得到目标产物;所述目标产物的结构式如下:
9.通过采用上述技术方案,直接采取卤代嘧啶作为原料与含有r2基团的联苯硼酸进
行偶联反应,除能有效提高原料转换率并保障产物的单一性,主产物收率为70-90%、纯度为95-99%,方便后处理外;还有利于含有r1基团的炔类化合物在钯配体和cui的催化下与主产物进一步反应得到次级产物,且所得到的次级产物只需一步几乎无损耗的氢化反应,即可得到目标产物,因而具有极高的效益。
10.优选的,所述s1的具体步骤如下:先取卤代嘧啶、含有r2基团的联苯硼酸、偶联助剂,溶于溶剂中,然后置换空气进行过夜反应,待tlc监测反应完毕后,经母液旋干、柱层析,即得主产物;其中偶联助剂为四三苯基膦钯、碳酸钾、三苯基膦、碳酸氢钠、醋酸钯中的一种或多种。
11.优选的,所述s1中的卤代嘧啶、含有r2基团的联苯硼酸、偶联助剂,其投料比eq 为(0.9-1.1):(0.9-1.1):(0.4-3.0)。
12.通过采用上述技术方案,以该投料比和反应条件偶联所得的主产物,其原料转换率较高的同时,产物较为单一,易于后处理作业的进行,所得主产物的收率为70-90%、纯度95-99%。
13.优选的,所述s2的具体步骤如下:先取s1中所得主产物、含有r1基团的炔类化合物、四三苯基膦钯、cui、溶于二甲基亚砜和三乙胺的混合溶剂中,然后置换空气并过夜反应,待tlc监测反应完毕后,经母液旋干、柱层析,即得次级产物。
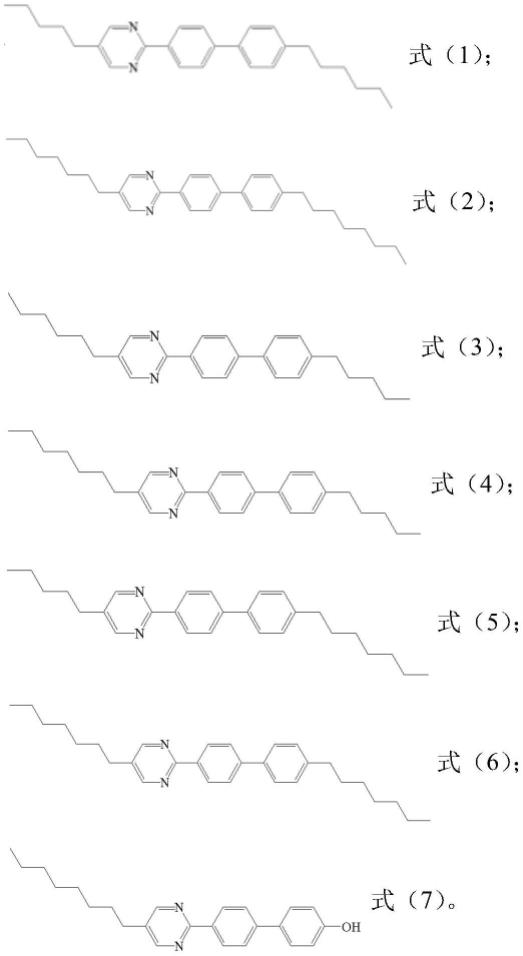
14.优选的,所述含有r1基团的炔类化合物、四三苯基膦钯和cui,其投料比eq为, (2.2-2.8):(0.03-0.08):(0.10-0.12)。
15.通过采用上述技术方案,以该投料比和条件进行的催化反应,其反应干净、基本无副产物产生的同时,收率较高,为80-95%,且所得到的次级产物仅需氢化反应,即能以近100%的转化率得到目标产物。
16.优选的,所述s3的具体步骤如下:取s2中所得次级产物和氢化助剂,搅拌溶于醇溶液,置换氢气并在氢气气氛下常温反应48
‑ꢀ
72h,待tlc监测反应完毕后,经母液旋干、柱层析,即得目标产物,其中氢化助剂为pd/c 或二氧化铂。
17.优选的,所述s3中的s2所得次级产物与氢化助剂的用量比为1:(0.1-0.3)。
18.通过采用上述技术方案,能以近100%的转化率得到目标产物,相比相关技术中通过r1的格式试剂直接与吡啶环上的卤素偶联,反应制得目标产物,收率显著提升,且产物易于分离。
19.优选的,所述目标产物包括如下化合物;
20.综上所述,本技术具有以下有益效果:1、本技术通过先将卤代嘧啶与含有r2基团的联苯硼酸进行偶联反应,再与含有r1基团的炔类化合物在钯配体和cui的催化下反应,最后经氢化反应,即可以较高的收率和纯度得到目标产物,且副产物单一且与产物极性相差较大,极大方便柱层析;2、本技术通过进一步优化偶联反应的投料比和条件,增加了反应原料的转换率,所得主产物的收率为70-90%、纯度95-99%,且所产物较为单一,易于后处理作业的进行; 3、本技术通过通过进一步优化催化反应的投料比和条件,保障了反应的高收率,为80
‑ꢀ
95%,且整体反应干净、基本无副产物产生,且所得产物有利于后续的氢化作业和后处理。
具体实施方式
以下结合实施例对本技术作进一步详细说明:本技术的各实施例及检测中所用的原料和设备,除下述特殊说明之外,其他均为市售:
旋转蒸发仪,型号ika rv10,采购自德国ika公司;检测设备:varian 400核磁共振波谱仪,采购自美国varian公司。
22.agilent 7890b-5977a气质联用仪,采购自美国安捷伦公司;agilent 6120液质联用仪,采购自美国安捷伦公司;实验所涉及的化学试剂及溶剂均采购自泰坦科技股份有限公司旗下阿达玛斯试剂品牌。实施例
23.实施例1一种嘧啶联苯芳香环化合物,其结构式为:
24.上述嘧啶联苯芳香环化合物的合成反应方程式如下:
25.上述合成反应的具体步骤为:s1、先取76.5g(1.0eq)5-溴-2-碘嘧啶、72g(1.0eq)含有r2基团的联苯硼酸、31g(0.1eq)四三苯基膦钯和92.8g(2.5eq)碳酸钾,溶于336ml的水和1.7l的1,4-二氧六环的混合液中;其中含有r2基团的联苯硼酸为(4-戊基[1,1-联苯]-4-基)硼酸;偶联助剂为四三苯基膦钯和碳酸钾;然后置换空气,600r/min搅拌10min,100℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得71.7g的主产物c
21h21
brn2,收率 70%,纯度96%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.46(d,j=8.5hz,2h), 7.72(d,j=8.5hz,2h),7.59(d,j=8.1hz,2h),7.30(d,j=8.1hz,2h),2.70
–
2.56(m,2h), 1.62(d,j=24hz,2h),1.34(dd,j=8.6,5.4hz,6h),0.91(t,j=6.7hz,3h)];s2、取38.1g的s1中所得主产物、21.5g(2.5eq)含有r1基团的炔类化合物、5.78g(0.05eq) 四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-己炔;
70%,纯度96%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.45(d,j=8.0hz,2h), 7.72(d,j=8.3hz,2h),7.60(d,j=7.0hz,2h),7.28(d,j=8.0hz,2h),2.65(t,j=7.7hz,2h), 1.63(d,j=6.5hz,2h),1.38
–
1.22(m,10h),0.90(t,j=6.8hz,3h)];s2、先取38.1g的s1中所得主产物、25.2g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-庚炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得34.1g的次级产物c
28h32
n2,收率86%,纯度98%;次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.76(s,2h),8.46(d,j=8.4hz,2h), 7.71(d,j=8.5hz,2h),7.58(d,j=8.1hz,2h),7.31
–
7.21(m,2h),3.77
–
3.66(m,2h), 2.69
–
2.57(m,2h),2.47(t,j=7.1hz,2h),1.68
–
1.61(m,4h),1.54
–
1.44(m,2h),1.32
ꢀ–
1.20(m,14h),0.99
–
0.83(m,6h)];s3、取10g的s2中所得次级产物和2g的pd/c(湿基,10%pd/c,wetted with ca.55%water),溶于甲醇溶液,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得9.69g的目标产物,收率96%,纯度99%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.56(s,2h),8.38(d,j=8.4hz,2h), 7.64(d,j=8.5hz,2h),7.52(d,j=8.1hz,2h),7.21(s,2h),2.56(dd,j=15.7,8.4hz,4h),1.55 (d,j=0.7hz,4h),1.23(d,j=25.6hz,18h),0.85
–
0.74(m,6h)]。
[0029]
实施例3一种嘧啶联苯芳香环化合物,其结构式为:
[0030]
上述嘧啶联苯芳香环化合物的合成反应方程式如下:
[0031]
上述合成反应的具体步骤为:s1、先取76.5g(1.0eq)5-溴-2-碘嘧啶、79.5g(1.0eq)含有r2基团的联苯硼酸、31g
(0.1eq) 四三苯基膦钯和92.8g(2.5eq)碳酸钾,溶于336ml的水和1.7l的1,4-二氧六环的混合液中;其中含有r2基团的联苯硼酸为(4-庚基[1,1-联苯]-4-基)硼酸;偶联助剂为四三苯基膦钯和碳酸钾;然后置换空气,600r/min搅拌10min,100℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得82.4g的主产物c
23h25
brn2,收率 75%,纯度95%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.46(d,j=8.5hz,2h), 7.72(d,j=8.5hz,2h),7.59(d,j=8.1hz,2h),7.29(d,j=8.1hz,2h),2.72
–
2.58(m,2h), 1.63(d,j=22.2hz,2h),1.35(dd,j=8.6,5.4hz,4h),0.91(t,j=6.7hz,3h)];s2、先取40.9g的s1中所得主产物、28.9g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-辛炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得37.2g的次级产物c
31h38
n2,收率85%,纯度97%;次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.78(s,2h),8.47(d,j=8.2hz,2h), 7.72(d,j=8.1hz,2h),7.60(d,j=7.9hz,2h),7.27(dd,j=9.1,4.3hz,2h),2.66(t,j=7.7hz, 2h),2.48(t,j=7.1hz,2h),1.86
–
1.57(m,4h),1.57
–
1.45(m,2h),1.40
–
1.34(m,4h), 0.94(dt,j=10.8,5.7hz,6h)];s3、取10g的s2中所得次级产物和2g的pd/c(湿基,10%pd/c,wetted with ca.55%water),溶于甲醇溶液,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得9.69g的目标产物,收率96%,纯度98%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.64(s,2h),8.46(d,j=8.4hz,2h), 7.72(d,j=8.4hz,2h),7.60(d,j=8.0hz,2h),7.34
–
7.23(m,2h),2.64(dd,j=15.6,8.0hz, 4h),1.86
–
1.61(m,4h),1.35(dd,j=9.5,5.8hz,10h),0.90(d,j=5.3hz,6h)]。
[0032]
实施例4一种嘧啶联苯芳香环化合物,其结构式为:
[0033]
上述嘧啶联苯芳香环化合物的合成反应方程式如下:
[0034]
上述合成反应的具体步骤为:s1、先取76.5g(1.0eq)5-溴-2-碘嘧啶、79.5g(1.0eq)含有r2基团的联苯硼酸、31g(0.1eq) 四三苯基膦钯和92.8g(2.5eq)碳酸钾,溶于336ml的水和1.7l的1,4-二氧六环的混合液中;其中含有r2基团的联苯硼酸为(4-庚基[1,1-联苯]-4-基)硼酸;偶联助剂为四三苯基膦钯和碳酸钾;然后置换空气,600r/min搅拌10min,100℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得80.2g的主产物c
23h25
brn2,收率 73%,纯度96%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.46(d,j=8.5hz,2h), 7.72(d,j=8.5hz,2h),7.59(d,j=8.1hz,2h),7.29(d,j=8.1hz,2h),2.72
–
2.58(m,2h), 1.63(d,j=22.2hz,2h),1.35(dd,j=8.6,5.4hz,4h),0.91(t,j=6.7hz,3h)];s2、先取40.9g的s1中所得主产物、17.86g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-戊炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得31.7g的次级产物c
28h32
n2,收率80%,纯度98%;次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.78(d,j=1.5hz,2h),8.51
–ꢀ
8.43(m,2h),7.76
–
7.66(m,2h),7.59(d,j=8.2hz,2h),7.27(dd,j=7.9,4.7hz,2h),2.65(t, j=7.8hz,2h),2.47(t,j=7.2hz,2h),1.64(dd,j=20.1,13.3hz,4h),1.48
–
1.19(m,8h), 0.99
–
0.83(m,6h)];s3、取10g的s2中所得次级产物和2g的pd/c(湿基,10%pd/c,wetted with ca.55%water),溶于甲醇溶液,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得9.6g的目标产物,收率95%,纯度98%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.64(s,2h),8.47(d,j=8.3hz,2h), 7.72(d,j=8.4hz,2h),7.60(d,j=8.0hz,2h),7.28(d,j=8.0hz,2h),2.64(dd,j=16.7,8.8 hz,4h),1.85
–
1.52(m,4h),1.47
–
1.19(m,12h),0.90(q,j=6.9hz,6h)]。
[0035]
实施例5
一种嘧啶联苯芳香环化合物,其结构式为:
[0036]
上述嘧啶联苯芳香环化合物的合成反应方程式如下:
[0037]
上述合成反应的具体步骤为:s1、先取76.5g(1.0eq)5-溴-2-碘嘧啶、75.8g(1.0eq)含有r2基团的联苯硼酸、31g(0.1eq) 四三苯基膦钯和92.8g(2.5eq)碳酸钾,溶于336ml的水和1.7l的1,4-二氧六环的混合液中;其中含有r2基团的联苯硼酸为(4-己基[1,1-联苯]-4-基)硼酸;偶联助剂为四三苯基膦钯和碳酸钾;然后置换空气,600r/min搅拌10min,100℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得82.8g的主产物c
22h23
brn2,收率 78%,纯度97%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.46(d,j=8.3hz,2h), 7.72(d,j=8.3hz,2h),7.59(d,j=8.0hz,2h),7.28(d,j=8.0hz,2h),2.66(t,j=7.7hz,2h), 1.65(d,j=6.9hz,2h),1.40
–
1.24(m,8h),0.89(t,j=6.6hz,3h)];s2、先取39.5g的s1中所得主产物、17.86g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-戊炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得30.96g的次级产物c
27h30
n2,收率81%,纯度97%;次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.78(s,2h),8.47(d,j=8.4hz,2h), 7.72(d,j=8.4hz,2h),7.59(d,j=8.0hz,2h),7.34
–
7.23(m,2h),2.71
–
2.59(m,2h), 2.46(t,j=7.0hz,2h),1.67(dp,j=14.7,7.4hz,4h),1.32(dd,j=16.2,6.6hz,8h),1.08(t,j= 7.4hz,3h),0.89(t,j=6.7hz,3h)];s3、取10g的s2中所得次级产物和2g的pd/c(湿基,10%pd/c,wetted with ca.55%water),溶于甲醇溶液,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;
待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得9.7g的目标产物,收率96%,纯度97%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.64(s,2h),8.46(d,j=8.4hz,2h), 7.77-7.68(m,2h),7.60(d,j=8.0hz,2h),7.28(d,j=8.1hz,2h),2.64(dd,j=15.9,8.7hz, 4h),1.72-1.63(m,4h),1.43-1.22(m,12h).0.90(dt,j=9.8,6.4hz,6h)]。
[0038]
实施例6一种嘧啶联苯芳香环化合物,其结构式为:
[0039]
上述嘧啶联苯芳香环化合物的合成反应方程式如下:
[0040]
上述合成反应的具体步骤为:s1、先取76.5g(1.0eq)5-溴-2-碘嘧啶、83.3g(1.0eq)含有r2基团的联苯硼酸、31g(0.1eq) 四三苯基膦钯和92.8g(2.5eq)碳酸钾,溶于336ml的水和1.7l的1,4-二氧六环的混合液中;其中含有r2基团的联苯硼酸为(4-辛基[1,1-联苯]-4-基)硼酸;偶联助剂为四三苯基膦钯和碳酸钾;然后置换空气,600r/min搅拌10min,100℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得85.3g的主产物c
24h27
brn2,收率 75%,纯度95%;主产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.84(s,2h),8.46(d,j=8.3hz,2h), 7.72(d,j=8.3hz,2h),7.59(d,j=8.0hz,2h),7.28(d,j=8.0hz,2h),2.66(t,j=7.7hz,2h), 1.65(d,j=6.9hz,2h),1.40
–
1.24(m,8h),0.89(t,j=6.6hz,3h)];s2、先取42.3g的s1中所得主产物、25.2g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-庚炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得35.1g的次级产物c
31h38
n2,收率80%,纯度95%;
次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.77(s,2h),8.47(d,j=8.4hz,2h), 7.72(d,j=8.5hz,2h),7.59(d,j=8.1hz,2h),7.33
–
7.23(m,2h),3.78
–
3.65(m,2h), 2.71
–
2.59(m,2h),2.47(t,j=7.1hz,2h),1.69
–
1.62(m,4h),1.52
–
1.42(m,2h),1.33
ꢀ–
1.20(m,14h),0.99
–
0.83(m,6h)];s3、取10g的s2中所得次级产物和2g的pd/c(湿基,10%pd/c,wetted with ca.55%water),溶于甲醇溶液,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得9.6g的目标产物,收率95%,纯度95%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.63(s,2h),8.46(d,j=8.2hz,2h), 7.72(d,j=8.3hz,2h),7.59(d,j=8.0hz,2h),7.27(d,j=7.8hz,2h),2.64(dd,j=16.6,8.0 hz,4h),1.66(s,4h),1.43-1.22(m,18h).0.88(t,j=6.1hz,6h)]。
[0041]
实施例7一种嘧啶联苯芳香环化合物,其结构式为:
[0042]
上述嘧啶联苯芳香环化合物的合成反应方程式如下:
[0043]
上述合成反应的具体步骤为:s1、先取4.02g(1.0eq)5-溴-2-碘嘧啶、4.13g(1.03eq)含有r2基团的联苯硼酸、 344mg(0.094eq)三苯基膦、1.65g(1.4eq)碳酸氢钠和180mg醋酸钯(0.057eq),溶于300ml 水与600ml异丙醇的混合液中;其中含有r2基团的联苯硼酸为4'-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)联苯-4-醇;偶联助剂为三苯基膦、碳酸氢钠和醋酸钯;然后置换氩气,600r/min搅拌10min,98℃过夜反应14h,待tlc监测反应完毕后,经旋转蒸发仪母液旋干、柱层析(tlc:pe/ea=10),即得3.23g的主产物c
16h11
brn2o,收率 70%,纯度95%;
主产物的核磁谱图读数为[1h nmr(400mhz,dmso)δ9.68(s,1h),9.05(s,2h),8.37(d,j= 8.3hz,2h),7.74(d,j=8.3hz,2h),7.59(d,j=8.4hz,2h),6.86(d,j=8.4hz,2h)];s2、先累计取32.7g的s1中所得主产物、28.9g(2.5eq)含有r1基团的炔类化合物、 5.78g(0.05eq)四三苯基膦钯、2g(0.11eq)cui、溶于200ml的二甲基亚砜和100ml的三乙胺的混合溶剂中,然后置换空气,600r/min搅拌10min,70℃过夜反应14h;其中含有r1基团的炔类化合物为1-辛炔;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),即得28.5g的次级产物c
24h24
n2o,收率80%,纯度95%;次级产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.78(s,2h),8.44(d,j=6.4hz,2h), 7.66(d,j=6.4hz,2h),7.55(d,j=6.4hz,2h),6.93(d,j=7.0hz,2h),2.47(t,j=6.1hz,2h), 1.69
–
1.31(m,8h),0.91(d,j=6.8hz,3h)];s3、取20g的s2中所得次级产物和2g的二氧化铂,溶于无水乙醇,600r/min搅拌10min,置换氢气并在氢气气氛下常温反应48h;待tlc监测反应完毕后,经母液旋干、柱层析(tlc:pe/ea=10),硅藻土过滤pd/c,母液旋干,即得19.2g的目标产物,收率95%,纯度95%;目标产物的核磁谱图读数为[1h nmr(400mhz,cdcl3)δ8.65(s,2h),8.43(d,j=6.2hz,2h), 7.66(d,j=6.5hz,2h),7.54(d,j=6.6hz,2h),6.92(d,j=6.3hz,2h),6.21(s,1h),2.63(t,j= 7.1hz,2h),1.65(d,j=7.5hz,2h),1.41-1.23(m,10h).0.88(t,j=6.6hz,3h)]。
[0044]
对上述实施例1-7中所得产物的收率和纯度进行检测,检测数据如下:表:实施例1-7和对比例1中目标产物的收率和纯度由上表中数据可知,实施例1-7中,目标产物嘧啶联苯芳香环化合物的纯度均在95%以上,收率可达95-98%,可见本技术直接采取卤代嘧啶作为原料与含有r2基团的联苯硼酸进行偶联反应,除能有效提高原料转换率并保障产物的单一性,主产物收率为70-90%、纯度为95-99%,方便后处理外;还有利于含有r1基团的炔类化合物在钯配体和cui的催化下与主产物进一步反应得到次级产物,且所得到的次级产物只需一步几乎无损耗的氢化反应,即可得到目标产物。
[0045]
本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1