一种西他列汀中间体的制备方法与流程
1.本发明涉及一种西他列汀中间体的制备方法,属于药物中间体合成技术领域。
背景技术:
2.西他列汀(sitagliptin),由美国默克公司研制开发,是一种二肽基酶-4(dpp-4)抑制剂,其活性成分为(3r)-3-氨基-1-[3-(三氟甲基)-5,6-二氢-1,2,4-三唑并[4,3-a]吡嗪-7(8h)-基]-4-(2,4,5-三氟苯基)丁-1-酮,用于治疗2型糖尿病,不仅治疗效果好,而且在治疗的过程中也不会发生引起患者低血糖的风险。
[0003]
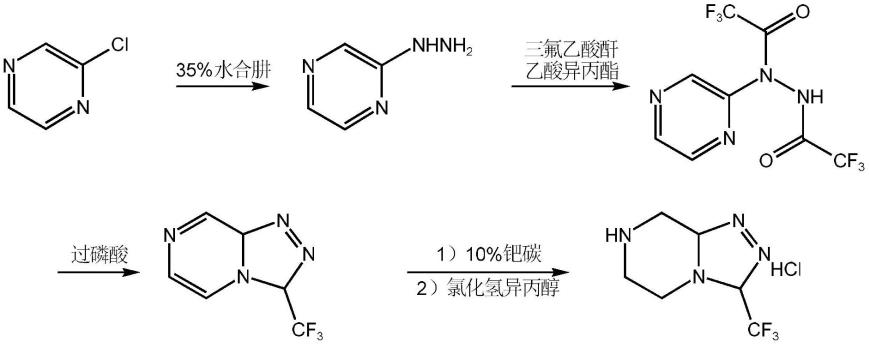
3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐是西他列汀合成中用到的关键中间体,其结构式如下所示:
[0004][0005]
关于该中间体的合成依照现有文献(wo2004103276a)说明,利用2-氯吡嗪先和水合肼、三氟乙酸酐反应,再经过磷酸脱水成环、钯碳催化加氢还原,盐酸成盐制备式ⅱ化合物,总收率为25%,该合成工艺的路线如下所示:
[0006][0007]
该方法中2-氯吡嗪、三氟乙酸酐、过磷酸、钯碳催化剂价格较高,且产品收率低,不利于成本控制;同时使用的三氟乙酸酐、过磷酸会产生大量的酸性废水,且较难处理,不利于环保;且钯碳催化加氢反应压力高,操作隐患大,也不利于安全和工业化生产。
[0008]
本发明中反应路线并未使用这些价格高昂的反应原料或催化剂,转而使用更改合成路线的方法将生产成本降低,并且排除了三氟乙酸酐、过磷酸等难以处理的容易造成污染的物质,同时对于可能危及安全的钯碳也弃之不用。
[0009]
又如cn102796104a中,用2-氯吡嗪与水合肼反应,添加氯苯和三氟乙酸酐加热100℃以上反应超100小时,消耗时间久,且产物纯度较低。
[0010]
因此,目前亟需寻找一种成本低、副产物少、产物率高,对环境友好的合成方法。
技术实现要素:
[0011]
本发明针对以上现有技术中存在的缺陷,提供一种西他列汀中间体的制备方法,解决的问题是如何实现降低成本,减少副产物产生,且提高产物收率和环保性好的路线。
[0012]
本发明的目的是通过以下技术方案得以实现的,一种西他列汀中间体的绿色合成方法,该方法包括以下步骤:
[0013]
s1:在醇溶剂中,将氯乙酸乙酯与1,2-乙二胺反应,生成2-哌嗪酮;
[0014]
s2:在醇溶剂中,将步骤s1中获取的中间化合物将2-哌嗪酮与三氟乙酸乙酯反应,生成n-三氟乙酰基哌嗪酮;
[0015]
s3:将步骤s2中获取的中间化合物n-三氟乙酰基哌嗪酮在boc保护下与五硫化二磷发生硫代反应,生成n-三氟乙酰基哌嗪硫酮;
[0016]
s4:在缚酸剂环境中,将步骤s3中获取的中间化合物n-三氟乙酰基哌嗪硫酮与水合肼进行反应,生成1-三氟乙酰肼基-2-哌嗪硫酮;
[0017]
s5:在醇溶剂中,将步骤s4中获取的中间化合物1-三氟乙酰肼基-2-哌嗪硫酮与盐酸反应发生环成盐反应,生成西他列汀中间体。
[0018]
其合成路线为:
[0019][0020]
本发明通过哌嗪酮与三氟乙酸乙酯发生反应生成n-三氟乙酸乙酯哌嗪酮,减少副产物的产生并且提高反应效率和产物生成率,同时反应环境变为醇溶剂,减少了对乙腈溶剂及醚类溶剂的需求,降低成本同时减小操作难度,减少了毒副作用便于后处理环境保护;生成产物n-三氟乙酸乙酯哌嗪酮在进行硫代反应后与水合肼可直接在缚酸剂下发生反应,无需添加三氯氧磷进行催化增加工作难度,减少了对环境的污染,并且反应更加稳定,转化率更高,反应过程中未使用大量催化物,生成废液及副产物较少,并且使中间化合物的产物生成率以及最终产物西他列汀中间体的纯度更高;最后只需要在醇溶剂中采用盐酸生成环成盐反应,无需使用钯碳对反应进行催化,减少了钯碳与氢接触反应导致压力升高的风向,不仅操作难度低,最终产物生成率高,且纯度也较高;并且本发明中反应路线短,减少了生成成本,由于整体反应环境中使用醇溶剂偏多,产生的副产物和废液便于后处理,利于减少对环境的污染,绿色生产。
[0021]
在上述西他列汀中间体的合成方法中,作为优选,醇溶剂可选用甲醇、乙醇和异丙醇中的一种或几种。采用醇溶剂具有安全性高,易于操作,对环境污染相对较少,能够进行回收利用,减少整体的生产成本等优点。作为优选,所述醇溶剂可选用乙醇,能够提高反应稳定性,更好的促进反应的进行,减少废液生成,降低后处理难度。
[0022]
在上述西他列汀中间体的合成方法中,作为优选,缚酸剂采用无机碱,无机碱选自
氢氧化钠、氢氧化钾其中的一种。使用缚酸剂可以在n-三氟乙酰基哌嗪硫酮与水合肼发生反应时,去除额外产生的的小分子盐,减少后处理难度,提高产物生成率。作为优选,所述缚酸剂可选用氢氧化钠,价格便宜且能够使反应更加稳定,提升产物纯度,减少毒副产物。
[0023]
在上述西他列汀中间体的合成方法中,作为优选,步骤s1反应过程中加入20%浓度醇钠稳定反应过程,醇钠可选用甲醇钠或乙醇钠其中一种。反应过程加入醇钠后,能够有效去除反应过程中生成的小分子物质,作为优选,所述醇钠选自乙醇钠,使转化效率更高,利于减少逆向反应,提高产物生成率。
[0024]
在上述西他列汀中间体的合成方法中,作为优选,步骤s2中反应在醇溶液与非水溶性溶剂双重环境中发生。使用醇溶剂与非水溶性双重环境,反应效率更高,同时非水溶性溶剂有利于中间化合物的生成以及后续反应的处理。作为优选,所述非水溶性溶剂可选用甲苯,优先选用沸点在85℃~95℃的非水溶性溶剂,醇溶剂与非水溶性溶剂配比为1:4~5。
[0025]
在上述西他列汀中间体的合成方法中,作为优选,步骤s4中反应在脱水环境下发生。反应过程中会生成小分子水,脱水能够提高反应转化率,作为优选,可选用5a分子筛进行脱水。
[0026]
在上述西他列汀中间体的合成方法中,作为优选,步骤s5中发生环成盐反应的温度为48~60℃,加入醇溶剂质量为1-三氟乙酰肼基-2-哌嗪硫酮的质量的4-8倍,便于反应能够稳定发生,提高反应效率,增加产物收率,作为优选,反应在56℃时反应最为稳定,反应效率效率最高。
[0027]
本发明的西他列汀中间体的合成方法的反应方程式如下所示:
[0028][0029]
综上所述,本发明与现有技术相比,具有以下优点:
[0030]
1.本发明反应环境采用醇溶剂环境,无需过多采用其它有毒有害溶剂,生成废液及其它副产物后处理方便,对环境污染小,得到的最终产物收率较高和纯度也较高,并且反应过程无需钯碳催化,降低了风险,易于人员操作,安全性较高,相比于其它反应路线产生风险概率更小价格较低,容易控制生产成本。
[0031]
2.本发明中间化合物的产物生成率更高,尤其是1-三氟乙酰肼基-2-哌嗪硫酮生成率和纯度较高,生成率≥80%,纯度≥98%。
具体实施方式
[0032]
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
[0033]
实施例1
[0034]
第一步:取1,2-乙二胺30.05g(0.50mol)溶于150ml乙醇中,搅拌均匀,降温至5℃,控制温度缓慢滴加氯乙酸乙酯61.28g(0.50mol),控温在5℃,滴毕,搅拌5h,再缓慢滴加含量20%的乙醇钠的乙醇溶液(含乙醇钠34.03g,0.50mol),控温≤25℃,滴毕,保温搅拌1h,反应结束后,过滤,收集滤液进行减压蒸馏脱除溶溶剂,再向浓缩物中加入丙酮进行重结晶处理,得到相应的产物湿品,烘干,得到2-哌嗪酮44.65g(0.446mol),收率为89.2%,纯度为99.1%。
[0035]
第二步:取2-哌嗪酮50.06g(0.50mol)溶于乙醇500ml,缓慢滴入三氟乙酸乙酯71.04g(0.50mol),控温5℃,滴毕,缓慢升温至25℃搅拌反应4h,加入10g无水硫酸钠干燥2h,抽滤,收集滤液进行减压蒸馏脱除溶剂,同时可收集蒸出的乙醇,最终得到中间物n-三氟乙酰基哌嗪酮,反应结束后在中间物中加入100ml水,搅拌50min,静置分液,收集有机层,水层使用50ml甲苯提取一次,合并有机相获得含有中间物n-三氟乙酰基哌嗪酮的料液。
[0036]
第三步:将上步所得含有中间物n-三氟乙酰基哌嗪酮的料液中滴加含有boc酸酐81.5g的丙酮溶液750ml,室温搅拌,过滤,蒸除丙酮,有机相转入三口瓶中,滴加五硫化二磷111.2g,保温搅拌2h,再用乙酸乙酯萃取,获得n-三氟乙酰基哌嗪硫酮料液。
[0037]
第四步:将上步得到的料液转至溶于500ml洁净的三口瓶中,加入20g氢氧化钠,搅拌均匀,室温下滴加80%水合肼31.29g(0.50mol),滴毕缓慢升温至56℃,保温搅拌反应4h,反应结束后,降温至室温,静置、分液,收集有机相,有机相转入500ml三口瓶中,再加入50g的5a分子筛,然后升温至56℃进行搅拌充分反应3h脱水生成1-三氟乙酰肼基-2-哌嗪硫酮,反应完毕后,抽滤,收集的有机相进行减压蒸馏脱溶,加入200ml的甲基叔丁基醚在室温条件下进行打浆处理2h,然后,缓慢降温至5℃充分析晶,抽滤,得到湿品中间产物1-三氟乙酰肼基-2-哌嗪硫酮,再在55℃的条件下进行真空干燥,得干品中间产物1-三氟乙酰肼基-2-哌嗪硫酮73.76g,以三氟乙酸乙酯计,收率为88.9%,纯度为98.8%。
[0038]
实施例2
[0039]
本实施例为3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐的合成;
[0040]
在1000ml的洁净的三口瓶中加入上述实施例3得到的1-三氟乙酰肼基-2-哌嗪硫酮72.35g(0.44mol)和无水乙醇600ml,然后,在室温下滴加浓度为36%的精制盐酸44.61g,滴毕后,将反应体系升温至56℃,并保温进行搅拌反应3h,反应结束后,缓慢降温至5℃进行充分析晶,抽滤,得最终产物湿品,烘干,得干品最终产物3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐86.5g,收率为86%,纯度为99.0%,外标含量为98%。
[0041]
实施例3
[0042]
第一步:取1,2-乙二胺30.05g(0.50mol)溶于150ml甲醇中,搅拌均匀,降温至5℃,控制温度缓慢滴加氯乙酸乙酯61.28g(0.50mol),控温在5℃,滴毕,搅拌5h,再缓慢滴加含量20%的甲醇钠的甲醇溶液(含甲醇钠27.01g,0.50mol),控温≤25℃,滴毕,保温搅拌1h,反应结束后,过滤,收集滤液进行减压蒸馏脱除溶溶剂,再向浓缩物中加入丙酮进行重结晶处理,得到相应的产物湿品,烘干,得到2-哌嗪酮44.05g(0.44mol),收率为88.0%,纯度为98.8%。
[0043]
第二步:取2-哌嗪酮50.06g(0.50mol)溶于甲醇500ml,缓慢滴入三氟乙酸乙酯71.04g(0.50mol),控温5℃,滴毕,缓慢升温至25℃搅拌反应4h,加入10g无水硫酸钠干燥1h,抽滤,收集滤液进行减压蒸馏脱除溶剂,同时可收集蒸出的甲醇,最终得到中间物n-三
氟乙酰基哌嗪酮,反应结束后在中间物中加入100ml水,搅拌45min,静置分液,收集有机层,水层使用50ml甲苯提取一次,合并有机相获得含有中间物n-三氟乙酰基哌嗪酮的料液。
[0044]
第三步:将上步所得含有中间物n-三氟乙酰基哌嗪酮的料液中滴加含有boc酸酐81.5g的丙酮溶液750ml,室温搅拌,过滤,蒸除丙酮,有机相转入三口瓶中,滴加五硫化二磷111.2g,保温搅拌2h,再用乙酸乙酯萃取,获得n-三氟乙酰基哌嗪硫酮料液。
[0045]
第四步:将上步得到的料液转至500ml洁净的三口瓶中,加入20g氢氧化钠,搅拌均匀,室温下滴加80%水合肼31.29g(0.50mol),滴毕缓慢升温至50℃,保温搅拌反应3h,反应结束后,降温至室温,静置、分液,收集有机相,有机相转入500ml三口瓶中,再加入55g的5a分子筛,然后升温至55℃进行搅拌充分反应3h脱水生成1-三氟乙酰肼基-2-哌嗪硫酮,反应完毕后,抽滤,收集的有机相进行减压蒸馏脱溶,向脱溶后的浓缩物加入200ml的甲基叔丁基醚在室温条件下进行打浆处理2h,然后,缓慢降温至5℃充分析晶,抽滤,得到湿品中间产物1-三氟乙酰肼基-2-哌嗪硫酮,再在50℃的条件下进行真空干燥,得干品中间产物1-三氟乙酰肼基-2-哌嗪硫酮73.4g,以三氟乙酸乙酯计,收率约为88.4%,纯度为98.4%。
[0046]
在1000ml的洁净的三口瓶中加入上述得到的1-三氟乙酰肼基-2-哌嗪硫酮73.4g(0.45mol)和无水乙醇500ml,然后,在室温下滴加浓度为36%的精制盐酸46g,滴毕后,将反应体系升温至60℃,并保温进行搅拌反应2h,反应结束后,缓慢降温至0℃进行充分析晶,抽滤,得最终产物湿品,烘干,得干品最终产物3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐82g,收率为81.5%,纯度为98.3%,外标含量为99%。
[0047]
实施例4
[0048]
第一步:取1,2-乙二胺30.05g(0.50mol)溶于150ml异丙醇中,搅拌均匀,降温至5℃,控制温度缓慢滴加氯乙酸乙酯61.28g(0.50mol),控温在5℃,滴毕,搅拌5h,再缓慢滴加含量20%的甲醇钠的甲醇溶液(含甲醇钠34.03g,0.50mol),控温≤25℃,滴毕,保温搅拌1h,反应结束后,过滤,收集滤液进行减压蒸馏脱除溶溶剂,再向浓缩物中加入丙酮进行重结晶处理,得到相应的产物湿品,烘干,得到2-哌嗪酮44.55g(0.45mol),收率为89.0%,纯度为98.0%。
[0049]
第二步:取2-哌嗪酮50.06g(0.50mol)溶于异丙醇500ml,缓慢滴入三氟乙酸乙酯71.04g(0.50mol),控温5℃,滴毕,缓慢升温至25℃搅拌反应4h,加入10g无水硫酸钠干燥1h,抽滤,收集滤液进行减压蒸馏脱除溶剂,同时可收集蒸出的乙醇,最终得到中间物n-三氟乙酰基哌嗪酮,反应结束后在中间物中加入100ml水,搅拌45min,静置分液,收集有机层,水层使用50ml甲苯提取一次,合并有机相获得含有中间物n-三氟乙酰基哌嗪酮的料液。
[0050]
第三步:将上步所得含有中间物n-三氟乙酰基哌嗪酮的料液中滴加含有boc酸酐81.5g的丙酮溶液750ml,室温搅拌,过滤,蒸除丙酮,有机相转入三口瓶中,滴加五硫化二磷111.2g,保温搅拌1h,再用乙酸乙酯萃取,获得n-三氟乙酰基哌嗪硫酮料液。
[0051]
第四步:将上步得到的料液转至500ml洁净的三口瓶中,加入20g氢氧化钠,搅拌均匀,室温下滴加80%水合肼31.29g(0.50mol),滴毕缓慢升温至50℃,保温搅拌反应3h,反应结束后,降温至室温,静置、分液,收集有机相,有机相转入500ml三口瓶中,再加入55g的5a分子筛,然后升温至55℃进行搅拌充分反应3h脱水生成1-三氟乙酰肼基-2-哌嗪硫酮,反应完毕后,抽滤,收集的有机相进行减压蒸馏脱溶,向脱溶后的浓缩物加入200ml的乙基叔丁基醚在室温条件下进行打浆处理2h,然后,缓慢降温至5℃进行充分析晶2h,抽滤,得到湿品
中间产物1-三氟乙酰肼基-2-哌嗪硫酮,再在50℃的条件下进行真空干燥,得干品中间产物1-三氟乙酰肼基-2-哌嗪硫酮72.4g,以三氟乙酸乙酯计,收率约为87.2%,纯度为98.4%。
[0052]
在1000ml的洁净的三口瓶中加入上述得到的1-三氟乙酰肼基-2-哌嗪硫酮72.4g(0.44mol)和无水乙醇400ml,然后,在室温下滴加浓度为36%的精制盐酸45g,滴毕后,将反应体系升温至48℃,并保温进行搅拌反应2h,反应结束后,缓慢降温至5℃进行充分析晶,抽滤,得最终产物湿品,烘干,得干品最终产物3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐83.5g,收率为81%,纯度为98.6%,外标含量为99%。
[0053]
实施例5
[0054]
本实施例为3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐的合成;
[0055]
在1000ml的洁净的三口瓶中加入上述实施例3得到的1-三氟乙酰肼基-2-哌嗪硫酮72.35g(0.44mol)和无水甲醇500ml,然后,在室温下滴加浓度为36%的精制盐酸46g,滴毕后,将反应体系升温至60℃,并保温进行搅拌反应3h,反应结束后,缓慢降温至0℃进行充分析晶,抽滤,得最终产物湿品,烘干,得干品最终产物3-三氟甲基-5,6,7,8-四氢-1,2,4-三唑[4,3-a]吡嗪盐酸盐82.5g,收率为80.5%,纯度为99.5%,外标含量为101%。
[0056]
实施例6
[0057]
第一步:取1,2-乙二胺36.06g(0.60mol)溶于150ml乙醇中,搅拌均匀,降温至0℃,控制温度缓慢滴加氯乙酸乙酯61.28g(0.50mol),控温在0℃,滴毕,搅拌5h,再缓慢滴加含量20%的乙醇钠的乙醇溶液(含乙醇钠34.03g,0.50mol),控温≤25℃,滴毕,保温搅拌2h,反应结束后,过滤,收集滤液进行减压蒸馏脱除溶溶剂,再向浓缩物中加入丙酮进行重结晶处理,得到相应的产物湿品,烘干,得到2-哌嗪酮43.7g(0.437mol),收率为87.2%,纯度为98.3%。
[0058]
第二步:取2-哌嗪酮60.07g(0.60mol)溶于乙醇500ml,缓慢滴入三氟乙酸乙酯85.24g(0.60mol),控温0℃,滴毕,缓慢升温至25℃搅拌反应6h,加入10g无水硫酸钠干燥2h,抽滤,收集滤液进行减压蒸馏脱除溶剂,同时可收集蒸出的乙醇,最终得到中间物n-三氟乙酰基哌嗪酮,反应结束后在中间物中加入100ml水,搅拌45min,静置分液,收集有机层,水层使用50ml甲苯提取一次,合并有机相获得含有中间物n-三氟乙酰基哌嗪酮的料液。
[0059]
第三步:将上步所得含有中间物n-三氟乙酰基哌嗪酮的料液中滴加含有boc酸酐81.5g的丙酮溶液750ml,室温搅拌,过滤,蒸除丙酮,有机相转入三口瓶中,滴加五硫化二磷111.2g,保温搅拌2h,再用乙酸乙酯萃取,获得n-三氟乙酰基哌嗪硫酮料液。
[0060]
第四步:将上步得到的料液转至溶于500ml洁净的三口瓶中,加入28.05g氢氧化钾,搅拌均匀,室温下滴加80%水合肼37.54g(0.60mol),滴毕缓慢升温至48℃,保温搅拌反应3h,反应结束后,降温至室温,静置、分液,收集有机相,有机相转入500ml三口瓶中,再加入50g的5a分子筛,然后升温至48℃进行搅拌充分反应3h脱水生成1-三氟乙酰肼基-2-哌嗪硫酮,反应完毕后,抽滤,收集的有机相进行减压蒸馏脱溶,加入200ml的甲基叔丁基醚在室温条件下进行打浆处理2h,然后,缓慢降温至0℃充分析晶,抽滤,得到湿品中间产物1-三氟乙酰肼基-2-哌嗪硫酮,再在48℃的条件下进行真空干燥,得干品中间产物1-三氟乙酰肼基-2-哌嗪硫酮73.1g,以三氟乙酸乙酯计,收率约为88.1%,纯度为98.4%。
[0061]
实施例7
[0062]
将n-三氟乙酰基哌嗪硫酮料液转至溶于500ml洁净的三口瓶中,加入28.05g氢氧
化钾,搅拌均匀,室温下滴加80%水合肼37.54g(0.60mol),滴毕缓慢升温至60℃,保温搅拌反应3h,反应结束后,降温至室温,静置、分液,收集有机相,有机相转入500ml三口瓶中,再加入50g的5a分子筛,然后升温至60℃进行搅拌充分反应3h脱水生成1-三氟乙酰肼基-2-哌嗪硫酮,反应完毕后,抽滤,收集的有机相进行减压蒸馏脱溶,加入200ml的甲基叔丁基醚在室温条件下进行打浆处理2h,然后,缓慢降温至0℃充分析晶,抽滤,得到湿品中间产物1-三氟乙酰肼基-2-哌嗪硫酮,再在60℃的条件下进行真空干燥,得干品中间产物1-三氟乙酰肼基-2-哌嗪硫酮73.51g,以三氟乙酸乙酯计,收率约为88.6%,纯度为98.5%。
[0063]
实施例8
[0064]
本实施例为现有技术公告文件cn102796104a生产实施例:
[0065]
第一步:反应器中加入乙醇和水合肼,加热至58℃,滴加2-氯吡嗪,控温60-61℃反应15小时,冷至0℃用氢氧化钠中和ph=6,20℃搅拌1小时后用二甲苯稀释,浓缩,残留物在20-30℃用二氯甲烷/异丙醇萃取,过滤,浓缩后用甲基叔丁基醚稀释,冷至0℃下继续搅拌2小时,过滤沉淀物,用冷甲基叔丁基醚洗涤,干燥(30℃/小于50mbar)得2,hplc纯度为93.30%;
[0066]
第二步:反应器中加入氯苯、三氟乙酸酐,冷却至0℃,激烈搅拌下分批加入2,控温3-5℃,加热至50℃,加7.0g(0.07mol)甲磺酸于混合反应液中,回流,蒸出三氟乙酸(bp 72-75℃),混合物在110℃反应42小时,100℃反应60小时,冷却至20℃,用乙酸钠作用减压浓缩至干,用冷24%-27%的氨水调节ph至12,过滤洗涤。分出有机相,用饱和氯化钠洗涤,干燥。用短硅胶柱过滤,用二氯甲烷洗涤,减压浓缩滤液得到4,hplc纯度为99.1%;
[0067]
第三步:不锈钢高压釜内氮气保护,加入10%钯碳及4的乙醇溶液,在23-25℃,4bar压力下氢化反应4.5小时,过滤洗涤,浓缩,残留物与氯化氢的乙醇溶液混合,混合液在25℃搅拌1小时,0℃搅拌2小时,-15℃下过夜,过滤分出沉淀物,用冷的乙醇/甲基叔丁基醚洗涤,干燥至恒重,得白色晶体,hplc纯度为99.3%,即为3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐。
[0068]
相比本技术方案,公告cn102796104a中反应过程所消耗的温度高时间长,消耗能量更多,产物纯度低,而且生产过程中高温对工作人员有安全隐患,不利于放大生产。
[0069]
本发明中所描述的具体实施例仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
[0070]
尽管对本发明已作出了详细的说明并引证了一些具体实施例,但是对本领域熟练技术人员来说,只要不离开本发明的精神和范围可作各种变化或修正是显然的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1