一种不对称氢化制备手性哌甲酯类化合物的方法与流程
1.本发明属于手性合成领域,具体涉及一种不对称氢化制备手性哌甲酯类化合物的方法。
背景技术:
2.过渡金属催化的不对称氢化将分子氢添加到前手性不饱和烯烃中,已被广泛用于构建光学纯精细化学品、农药和药物。与二取代和三取代烯烃相比,四取代烯烃的不对称氢化仍然是该领域的一大挑战。迄今为止,对空间位阻要求较低的氟代或甲基取代的四取代烯烃的不对称氢化也仅有几篇报道(如下所示)。然而,诸如官能团不兼容、反应活性差、对映选择性低以及对具有大空间位阻的底物无效等限制,仍然阻碍了不对称氢化在四取代烯烃中的应用。
[0003][0004]
哌甲酯(methylphenidate)属中枢兴奋剂,是临床治疗注意缺陷多动障碍的一线药物,通过促进多巴胺释放、减少多巴胺再摄取及抑制单胺氧化酶活性而起作用,可显著减少多动行为,增加注意力集中能力,有效改善和治疗注意力缺乏或多动综合症(adhd)。1944年,panizzon等报道合成哌甲酯的四种异构体 [(
±
)-erythro/(
±
)-threo)]混合物,并于1950年以商品名利他林(ritalin)在美国上市。
[0005][0006]
根据更进一步的临床研究表明,苏式构型(threo)对上述疾病才具有治疗作用,
(2r,2’r)-苏型哌甲酯更容易进入中枢神经系统,比其对映体更具药理活性。 d-苏型哌甲酯的缓释胶囊于2005年5月26日由novartis公司通过fda申请上市,商品名为focalin xr。
[0007]
d-苏型哌甲酯的合成根据目前的专利文献报道主要有如下几种方法:
[0008]
一、手性拆分方法:参考专利和文献wo 98/52921,1998;us patent 5,936,091,1999;tetrahedron:asymmetry 1998,9,2133。
[0009]
二、手性合成法:
[0010]
1)novartis公司报道了第一例手性全合成d-苏型哌甲酯的方法(j.org.chem. 1999,64,1750.),该方法利用光学纯的噁唑啉酮手性辅助基,与5-氯戊醛发生羟醛缩合反应得到构型单一的产物,再经保护、环化、去保护等步骤得到目标化合物,其合成路线如反应式i:
[0011][0012]
2)winkler和davies的研究小组报道了铑催化的不对称卡宾插入反应合成 d-苏型哌甲酯的方法(j.am.chem.soc.1999,121,6509;j.am.chem.soc.1999, 121,6511.)。该方法十分简洁的构建了哌甲酯结构,可以通过控制催化剂的构型获得不同构型的哌甲酯衍生物,其合成路线如反应式ii:
[0013][0014][0015]
3)matsumura小组报道了从保护的哌啶出发,先通过电化学氧化得到保护的2-甲氧基哌啶,再与苯乙酸的手性噁唑啉酮酰胺反应立体选择性构建哌甲酯衍生物,经去保护、皂化、酯化等步骤得到目标化合物(org.lett.1999,1,175; tetrahedron 2000,56,7411.)。其合成路线如反应式iii:
[0016][0017]
4)专利cn 102134208a报道了利用叔丁基亚磺酰胺与5-氯戊醛缩合得到叔丁基亚磺酰亚胺,然后与苯乙酸甲酯在强碱的条件下发生加成反应形成手性中间体,再通过去保护和关环反应得到d-苏型哌甲酯,光学纯度达到》97%ee。其合成路线如反应式iv:
[0018][0019]
5)perel的研究小组以d-哌啶酸为原料,经保护、偶联反应、wittig反应、硼氢化氧化、醇氧化、甲基化和酸化等步骤得到目标化合物(j.med.chem.1998, 41,591)。该方法以光学纯的底物,可诱导一系列右哌甲酯衍生物的制备,其合成路线如反应式
ⅴ
:
[0020][0021][0022]
综上所述,现有技术中多采用手性拆分和手性辅基诱导的合成方法,反应路径繁琐,成本过高,原料浪费严重,因此不利于大规模工业化生产。因此,亟需提供一种高效的手性哌甲酯化合物的全新合成工艺,以期通过廉价的试剂与温和的反应条件获得较高的产物收率和选择性,最终实现(r)-2-苯基-2-((r)-哌啶-2-基) 乙酸甲酯盐酸盐的大规模工业化生产。
技术实现要素:
[0023]
本发明要解决的技术问题是提供一种高效的手性哌甲酯化合物的全新合成工艺,以期通过廉价的试剂与温和的反应条件获得较高的产物收率和选择性,最终实现(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯盐酸盐的大规模工业化生产。
[0024]
本发明提供了一种不对称氢化制备手性哌甲酯类化合物的方法,其反应路线为:
[0025][0026]
具体地,化合物(i)在合适的溶剂中,加入手性双膦配体、过渡金属催化剂、布朗斯特酸以及添加剂,充入氢气进行不对称还原反应得到式(ii)所示的化合物。
[0027]
其中,化合物中r代表不同的取代基,为烷基、环烷基、芳烷基、杂环烷基;另外,上述烷基、环烷基、芳烷基、杂环烷基也具有取代基,星号(*)标记的碳原子表示手性碳原子。
[0028]
作为本发明的一种优选技术方案,所用手性双膦配体选自以下至少一种:
[0029][0030][0031]
优选为(r)-f-binaphane或其任意对映异构体。
[0032]
作为本发明的一种优选技术方案,所述过渡金属选自铱、铑、钌;其中,金属前体选自[ir(nbd)cl]2,[ir(nbd)2]x,[ir(cod)cl]2,[ir(cod)2]x,[rh(nbd)2]x, [rh(nbd)cl]2,rh(acac)(co)2,[rh(cod)cl]2,rh(ethylene)2(acac), [rh(ethylene)2cl]2,[rh(cod)2]x,rhcl(pph3)3,ru(aryl group)x2,rux2(cymene), rucl2(cod),(ru(cod)2)x,rux2(diphosphine),ru(arh)cl2, ru(cod)(methallyl)2;x表示负阴离子cl-,br-,i-,bf
4-,clo
4-,sbf
6-,pf
6-, tfo-,rcoo-,b(ar)
4-;金属优选铱金属,金属前体优选[ir(cod)cl]2。
[0033]
作为本发明的一种优选技术方案,所述反应是在溶剂中进行;所述溶剂选自甲醇、乙醇、异丙醇、甲基叔丁基醚、四氢呋喃、1,4-二氧六环、二氯甲烷、乙酸乙酯、正己烷、甲苯中的一种或任意比例的混合溶剂,优选为异丙醇。式(i) 所示的化合物与溶剂的用量比为1mmol:(10-20)ml,优选为1mmol:10ml。
[0034]
作为本发明的一种优选技术方案,使用的氢气压力为10-100bar的压力,优选30-60bar的氢气压力。
[0035]
作为本发明的一种优选技术方案,其特征在于,本发明所使用的反应温度选择20-100℃,其中,优选20-50℃。
[0036]
作为本发明的一种优选技术方案,本发明所使用的布朗斯特酸选自苯甲酸、甲酸、三氟甲磺酸、磷酸、樟脑磺酸、对甲苯磺酸、三氟乙酸,盐酸中的至少一种或任意比例的混合
物,优选为三氟乙酸。
[0037]
作为本发明的一种优选技术方案,本发明所使用的添加剂选自醋酸锌、三氟甲磺酸锌、氯化锌,碘,四叔丁基碘化铵,氯化铵,四异丙氧钛,三氟化硼,氯化锂、溴化锂、异丙基铝、氯化铝、氟化铝、溴化铝、碘化铝、三氟甲磺酸铝,氯化镁、氯化铟等,优选为三氯化铝。
[0038]
作为本发明的一种优选技术方案,催化剂的使用量根据氢化底物、反应条件及催化剂的种类而定,催化剂与底物的摩尔比范围为0.01mol%-10mol%,优选 0.11mol%-1mol%。
[0039]
作为本发明的一种优选技术方案,反应时间一般为0.5-100h,优选16-72h。
[0040]
本发明进一步提供了一种(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯盐酸盐的制备工艺,其特征在于,合成路线如下:
[0041][0042]
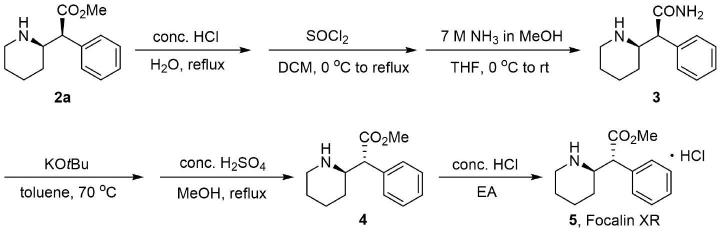
具体的,包括以下步骤:1)(s)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯(2a)与浓盐酸加热回流反应脱去甲酯,加入二氯亚砜活化后,经胺解得到甲酰胺取代的化合物3;2)化合物3在合适的溶剂中,和强碱反应在加热条件下异构化,再经水解甲酯化得到(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯(4);3)化合物4与盐酸成盐得到(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯盐酸盐(5)。
[0043]
本发明相对于现有技术具有以下有益效果:
[0044]
(1)本发明提供了一种手性哌甲酯化合物的全新合成工艺,反应具有高度的稳定性和反应活性,实现了优异的立体控制,可以得到大于90%对映选择性的哌甲酯中间体,且dr值大于20:1。
[0045]
(2)本发明中,不对称催化还原反应是关键步骤,使用优选的催化体系 ir/f-binaphane,不对称氢化反应具有非常高的反应活性,催化剂转化数(ton, turnover number)高达1500,并且能保持其优异的立体控制。
[0046]
(3)本发明的工艺具有较高的产物收率和选择性,成本低廉,易于放大,适用于大规模工业化生产,具有极高的工业化价值。
具体实施方式
[0047]
下面结合具体实施方式对本发明的技术方案进一步的说明和描述,但本发明并不限制于以下描述的具体实施例。
[0048]
实施例中未注明具体条件的实验方法,通常按照常规条件以及手册中所述的条件,或按照制造厂商所建议的条件;所用材料、试剂等,如无特殊说明,均可从商业途径得到。
[0049]
实施例1:(s)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0050][0051]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、(r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(23.1mg,0.1mmol)、三氯化铝(2.7mg,0.02mmol)、1ml无水异丙醇和11μl三氟乙酸。在30bar的氢气氛围下反应24h后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(21.4mg,收率:92%,92%ee,98:2dr)。
[0052]
产物为无色液体,[α]
25d
=-58.8(c0.85,ch3oh),hplc条件:chiralpakie柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:1.0ml/min,254nm,t1=9.8min,t2=10.1min(major),t3=13.2min。1hnmr(600mhz,cdcl3)δ=7.41(d,j=7.0hz,2h),7.34(t,j=7.2hz,2h),7.31
–
7.29(m,1h),3.65(s,3h),3.46(d,j=10.0hz,1h),3.10(t,j=10.0hz,1h),2.92(d,j=11.1hz,1h),2.50(t,j=11.2hz,1h),1.81
–
1.79(m,2h),1.56(s,1h),1.46
–
1.22(m,4h).
13
cnmr(151mhz,cdcl3)δ=173.0,136.1,128.9,128.7,127.8,59.0,58.3,51.9,47.0,31.1,25.8,24.5.hrms(esi)m/z:[m+h]
+
calcdforc
14h20
no
2+
=234.1489;found234.1486.
[0053]
实施例2:(s)-2-(3-氟苯基)-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0054][0055]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、(r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(24.9mg,0.1mmol)、三氯化铝(2.7mg,0.02mmol)、1ml无水异丙醇和11μl三氟乙酸。在30bar的氢气氛围下反应24h后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(23.3mg,收率:93%,93%ee,98:2dr)。
[0056]
产物为无色液体,[α]
25d
=-33.1(c1.20,ch3oh),hplc条件:chiralpakic柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:0.5ml/min,254nm,t1=13.6min(major),t2=14.7min,t3=17.8min.1hnmr(400mhz,cdcl3)δ=7.33
–
7.26(m,2h),7.18
–
7.16(m,2h),7.02
–
6.97(m,1h),3.66(s,3h),3.46(d,j=10.0hz,1h),3.07(td,j=10.1,2.1hz,1h),2.94(d,j=11.7hz,1h),2.52(td,j=11.5,2.8hz,1h),1.81
–
1.76(m,2h),1.60
–
1.56(m,1h),1.47
–
1.19(m,4h).
13
cnmr(101mhz,cdcl3)δ=172.6,163.0(d,j=246.9hz),138.5(d,j=7.3hz),130.2(d,j=8.3hz),124.6,115.5(d,j=21.9hz),114.9(d,j=21.0hz),59.0,58.0,52.0,47.0,31.0,25.7,24.4.
19
fnmr(376mhz,cdcl3)δ=-112.2.hrms(esi)m/z:[m+h]
+
calcdforc
14h19
fno
2+
=252.1394;found252.1391.
[0057]
实施例3:(s)-2-(3-氯苯基)-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0058][0059]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、 (r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(26.5mg,0.1mmol)、三氯化铝(2.7mg,0.02 mmol)、1ml无水异丙醇和11μl三氟乙酸。在30bar的氢气氛围下反应24h 后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(24.3mg,收率:91%,92%ee,99:1dr)。
[0060]
产物为无色液体,[α]
25d
=-48.2(c 1.26,ch3oh),hplc条件:chiralpak ie 柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:0.5ml/min,254nm, t1=16.7min,t2=17.2min(major).1h nmr(400mhz,cdcl3)δ=7.42(s,1h), 7.30
–
7.24(m,3h),3.66(s,3h),3.42(d,j=10.0hz,1h),3.07(td,j=10.1,2.1hz, 1h),2.93(d,j=11.7hz,1h),2.51(td,j=11.5,2.8hz,1h),1.81
–
1.75(m,2h), 1.59
–
1.55(m,1h),1.47
–
1.18(m,4h).
13
c nmr(101mhz,cdcl3)δ=172.5, 138.1,134.7,130.0,128.7,128.1,127.0,58.9,58.0,52.0,47.0,31.0,25.7,24.4. hrms(esi)m/z:[m+h]
+
calcd for c
14h19
clno
2+
=268.1096;found 268.1099.
[0061]
实施例4:(s)-2-(3-溴苯基)-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0062][0063]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、 (r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(30.9mg,0.1mmol)、三氯化铝(2.7mg,0.02 mmol)、1ml无水异丙醇和11μl三氟乙酸。在30bar的氢气氛围下反应24h 后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(28.6mg,收率:92%,91%ee,99:1dr)。
[0064]
产物为无色液体,[α]
25d
=-42.3(c 1.20,ch3oh),hplc条件:chiralpak ic 柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:0.5ml/min,254nm, t1=14.5min,t2=15.4min(major).1h nmr(400mhz,cdcl3)δ=7.58(t,j=1.7 hz,1h),7.43(ddd,j=7.9,1.9,1.0hz,1h),7.34(d,j=7.8hz,1h),7.21(t,j=7.8 hz,1h),3.66(s,3h),3.41(d,j=10.0hz,1h),3.06(td,j=10.1,2.1hz,1h),2.94 (d,j=11.7hz,1h),2.52(td,j=11.5,2.7hz,1h),1.81
–
1.74(m,2h),1.59
–
1.56 (m,1h),1.47
–
1.18(m,4h).
13
c nmr(101mhz,cdcl3)δ=172.5,138.4,131.6, 131.1,130.3,127.4,122.9,59.0,58.0,52.0,47.0,31.0,25.7,24.4.hrms(esi)m/z: [m+h]
+
calcd for c
14h19
brno
2+
=312.0594;found 312.0590.
[0065]
实施例5:(s)-2-(3-三氟甲基苯基)-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0066]
binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(23.7mg,0.1mmol)、三氯化铝(2.7mg,0.02mmol)、1ml无水异丙醇和11μl三氟乙酸。在30bar的氢气氛围下反应24h后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(21.5mg,收率:90%,89%ee,99:1dr)。
[0076]
产物为无色液体,[α]
25d
=-53.5(c0.78,ch3oh),hplc条件:chiralpakie柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:0.8ml/min,254nm,t1=14.7min,t2=15.2min(major),t3=20.1min.1hnmr(400mhz,cdcl3)δ=7.31(dd,j=4.9,3.0hz,1h),7.25(d,j=1.0hz,1h),7.16(dd,j=4.9,1.0hz,1h),3.67(s,3h),3.62(d,j=9.8hz,1h),3.01(td,j=10.1,2.3hz,1h),2.95(d,j=11.5hz,1h),2.52(td,j=11.4,2.7hz,1h),1.81
–
1.74(m,2h),1.58
–
1.56(m,1h),1.47
–
1.18(m,4h).
13
cnmr(101mhz,cdcl3)δ=172.7,136.4,127.2,126.2,123.4,59.1,53.7,51.9,47.0,30.9,25.8,24.4.hrms(esi)m/z:[m+h]
+
calcdforc
12h18
no2s
+
=240.1053;found240.1050.
[0077]
实施例8:(s)-2-(萘-1-基)-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0078][0079]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、(r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(28.1mg,0.1mmol)、三氯化铝(2.7mg,0.02mmol)、1ml无水异丙醇和11μl三氟乙酸。在60bar的氢气氛围下反应48h后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(26.0mg,收率:92%,96%ee,97:3dr)。
[0080]
产物为白色固体,[α]
25d
=-81.1(c0.85,ch3oh),hplc条件:chiralpakie柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:1.0ml/min,254nm,t1=10.5min,t2=12.4min(major),t3=14.5min.1hnmr(400mhz,cdcl3)δ=8.28(d,j=8.5hz,1h),7.86(d,j=8.1hz,1h),7.80(d,j=8.2hz,1h),7.73(d,j=7.2hz,1h),7.57
–
7.53(m,1h),7.49(t,j=7.7hz,2h),4.42(d,j=9.9hz,1h),3.61(s,3h),3.37(t,j=9.6hz,1h),2.86(d,j=10.0hz,1h),2.50(td,j=11.2,2.9hz,1h),1.96
–
1.84(m,2h),1.58
–
1.55(m,1h),1.50
–
1.26(m,4h).
13
cnmr(101mhz,cdcl3)δ=173.2,134.0,132.5,132.4,128.8,128.2,126.6,125.8,125.6,123.4,59.4,51.9,47.1,31.2,25.8,24.6.hrms(esi)m/z:[m+h]
+
calcdforc
18h22
no
2+
=284.1645;found284.1642.
[0081]
实施例9:(s)-2-苯基-2-((r)-哌啶-2-基)乙酸苄酯的合成
[0082][0083]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(0.3mg,0.5μmol)、(r)-f-binaphane(0.9mg,1.1μmol)和30μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(30.7mg,0.1mmol)、三氯化铝(2.7mg,0.02mmol)、1ml无水异丙醇和11μl三氟
乙酸。在30bar的氢气氛围下反应24h 后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(28.1mg,收率:91%,92%ee,98:2dr)。
[0084]
产物为无色液体,[α]
25d
=-16.8(c 1.15,ch3oh),hplc条件:chiralpakadh 柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:1.0ml/min,254nm, t1=9.9min,t2=10.2min(major),t3=15.2min.1h nmr(400mhz,cdcl3)δ= 7.42
–
7.40(m,2h),7.35
–
7.23(m,8h),5.09(dd,j=40.8,12.5hz,2h),3.52(d,j =10.0hz,1h),3.13(t,j=10.1hz,1h),2.92(d,j=11.3hz,1h),2.50(t,j=10.8 hz,1h),1.78
–
1.76(m,2h),1.57
–
1.55(m,1h),1.47
–
1.21(m,4h).
13
c nmr (101mhz,cdcl3)δ=172.4,135.9,135.8,128.9,128.7,128.5,128.1,127.9,66.4, 59.0,58.4,47.0,31.0,25.7,24.4.hrms(esi)m/z:[m+h]
+
calcd for c
20h24
no
2+
= 310.1802;found 310.1802.
[0085]
实施例10:(s)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯的扩大合成
[0086][0087]
在氩气氛围下,向氢化瓶中加入[ir(cod)cl]2(1.0mg,1.5μmol)、 (r)-f-binaphane(2.7mg,3.3μmol)和90μl无水二氯甲烷。反应室温搅拌30分钟后,依次加入四取代烯烃原料(1.04g,4.5mmol)、三氯化铝(120mg,0.9mmol)、 4.5ml无水异丙醇和0.5ml三氟乙酸。在30bar的氢气氛围下反应72h后,原料全部转化为产物。随后缓慢释放氢气,将反应的ph值调至10,用乙酸乙酯萃取,收集有机相用无水硫酸钠干燥后减压条件下除去溶剂,然后柱层析得到目标产物(0.96g,收率:92%,92%ee,98:2dr)。
[0088]
实施例11:(s)-2-苯基-2-((r)-哌啶-2-基)乙酰胺的合成
[0089][0090]
向一个50ml反应瓶中加入2a(466mg,2.0mmol),10ml水和3ml浓盐酸,回流条件下搅拌过夜。待反应体系冷却后,减压条件下除去溶剂,得到淡黄色粗产品直接用于下一步反应。
[0091]
在20ml反应瓶中将粗产品溶于无水二氯甲烷,在0℃摄氏度缓慢滴加二氯亚砜。搅拌半个小时后,反应体系缓慢升温至回流再搅拌2小时。减压条件下除去溶剂后将残留物溶于12ml四氢呋喃,随后在0℃缓慢滴加7m的氨甲醇溶液,反应体系升至室温后搅拌2小时。在0℃用水淬灭,加入3ml 5m盐酸溶液搅拌1小时。萃取后收集水相,用30%的氢氧化钠水溶液将ph值调至12 再用乙酸乙酯萃取。收集有机相在减压条件下除去溶剂,经过柱层析就可以得到产物3(327mg,产率:75%)
[0092]
产物为白色固体。1h nmr(400mhz,cdcl3)δ=7.40
–
7.26(m,5h),6.77(s, 1h),5.51(s,1h),3.30(d,j=7.3hz,1h),3.13
–
3.08(m,1h),2.99
–
2.95(m,1h), 2.55(td,j=12.1,2.8hz,1h),1.86
–
1.79(m,2h),1.57
–
1.54(m,1h),1.46
–
1.27 (m,3h),1.21
–
1.14(m,1h).
13
c nmr(101mhz,cdcl3)δ=174.6,136.4,128.8, 128.8,127.7,58.7,58.4,47.0,30.9,26.1,
24.6.hrms(esi)m/z:[m+h]+calcdforc
13h19
no
2+
=219.1492;found219.1492.
[0093]
实施例12:(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯的合成
[0094][0095]
在氩气氛围下,向一个50ml反应瓶中加入3(280mg,1.3mmol),叔丁醇钾(291mg,2.6mol)和15ml甲苯,反应在70℃回流条件下搅拌24小时。在0℃用水淬灭,加入2ml5m盐酸溶液搅拌1小时。萃取后收集水相,用30%的氢氧化钠水溶液将ph值调至12再用乙酸乙酯萃取。收集有机相在减压条件下除去溶剂,得到白色固体。
[0096]
将上述白色固体溶于3ml甲醇中,向体系中加入0.5ml浓硫酸,反应在75℃下搅拌48小时。待反应冷却后减压条件下除去溶剂,在0℃加入5ml水和5ml乙酸异丁酯。用饱和碳酸钠水溶液将ph值调至12,再用乙酸乙酯萃取。收集有机相在减压条件下除去溶剂,得到产物4(198mg,0.85mmol,收率:66%,90%ee,96:4dr)。
[0097]
产物为无色液体。[α]
23d
=+74.1(c1.0,ch3oh);hplc条件:chiralpakie柱,正己烷(包含0.05%三氟乙酸)/异丙醇=98/2,流速:1.0ml/min,254nm,t1=10.5min,t2=12.9min(major),t3=14.8min.1hnmr(400mhz,cdcl3)δ=7.33
–
7.24(m,5h),3.64(s,3h),3.44(d,j=10.1hz,1h),3.11
–
3.05(m,2h),2.70(td,j=11.9,2.8hz,1h),1.95(s,1h),1.70
–
1.66(m,1h),1.59
–
1.56(m,1h),1.43
–
1.32(m,1h),1.28
–
1.17(m,2h),1.00
–
0.91(m,1h).
13
cnmr(101mhz,cdcl3)δ=173.8,136.4,128.6,128.5,127.5,58.99,58.7,51.9,46.8,29.9,26.1,24.3.hrms(esi)m/z:[m+h]+calcdforc
14h20
no
2+
=234.1489;found234.1488.
[0098]
实施例13:(r)-2-苯基-2-((r)-哌啶-2-基)乙酸甲酯盐酸盐的合成
[0099][0100]
将4(152mg,0.65mmol)溶于3ml乙酸乙酯中,向体系中加入0.5ml4m盐酸二氧六环溶液,搅拌半个小时。将反应体系过滤,收集滤饼,减压条件下干燥得到产物5(176mg,0.65mmol,收率:99%)。
[0101]
产物为白色固体。[α]
23d
=+78.8(c1.0,ch3oh).1hnmr(400mhz,cdcl3)δ=10.37(brs,1h),8.90(brs,1h),7.37
–
7.27(m,5h),4.31(d,j=10.2hz,1h),3.83(s,3h),3.73
–
3.63(m,2h),2.92(t,j=12.3hz,1h),2.16
–
2.06(m,1h),1.84
–
1.69(m,3h),1.41
–
1.33(m,2h).
13
cnmr(101mhz,cdcl3)δ=172.0,134.1,129.2,128.4,128.4,59.0,53.9,53.4,45.6,25.9,22.6,21.9.hrms(esi)m/z:[m
–
cl
–
]
+
calcdforc
14h20
no
2+
=234.1489;found234.1489.
[0102]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1