氨基硅油的制备方法与流程
1.本发明涉及化学分析技术领域,具体为一种氨基硅油的制备方法。
背景技术:
2.氨基硅油作为一种功能性硅油,是有机硅化合物中应用最广的产品之一,具有吸附、顺滑、润滑、成膜、消泡、着色等多项优良性能,被广泛应用于织物整理、皮革后处理、个人护理用品等诸多领域。
3.目前工业上常用的合成方法以本体聚合和乳液聚合为主。但应用乳液聚合法合成得到的氨基硅油不可避免会有少量硅氧烷单体残留,以其配置乳液应用时易发生破乳现象从而形成硅斑等问题,难以去除影响产品质量。本体聚合法其工艺简单、反应过程容易控制,适合大规模生产,目前多采用偶联剂直接与dmc反应的共聚合成法。由于该方法涉及硅氧烷环体的开环、偶联剂的缩聚及彼此共聚等多种机制,易造成反应体系中存在缩合出的醇类化合物,同时偶联剂水解不完全使得其对于目标产物的转化率不高,影响最终产品性能。
4.cn108676165a公开了一种氨基硅油原油的制备工艺,其通过分别将二甲基硅氧烷混合环体与封头剂、偶联剂与固体催化剂预混得到预混料,随后将两种预混料在反应容器中混合缩聚,最后经过脱低得到氨基硅油。其获得的氨基硅油原料转化率高达93.55%,但是,该方法中需要对原料进行预混,同时缩聚反应过程长达12~20小时,其工艺步骤繁琐,周期长,能耗高,并不利于实际生产应用。
5.cn108912333a公开了一种高透过率氨基硅油的制备方法,包括一个制备偶联剂水解物的步骤和一个制备氨基硅油的步骤。首先将八甲基环四硅氧烷脱水;然后常压加入催化剂溶解;再减压加入偶联剂水解物混合;接着减压加入封端剂反应再升温使得催化剂失活;最后升温至去除低分子化合物;静置冷却即可制得高透过率氨基硅油。其方法制备得到氨基硅油氨基分布有所提高,但是,偶联剂在低温无催化剂的条件下,无法保证水解完全,同时偶联剂的水解物间可发生缩聚反应,进而降低了氨基的均匀分布程度。
6.cn112280041a公开了一种有机硅环体含量低的氨基硅油的制备方法,将羟基封端的聚二甲基硅氧烷、具有硅烷氧基的氨基单体和封端剂在50~100℃的温度下进行搅拌混合;向混合均匀的物料中加入水进行水解,通入氮气排除水分和醇;向水解后的物料中加入碱性催化剂,在50~110℃的温度下进行聚合反应;最后对反应物料进行脱低,得到低环体含量的氨基硅油。但是,其所用主要原料为低粘度线性体,其获取难度高、成本高。同时由于线性体两端为羟基,易发生缩合,粘度不易控制,需要额外加入封端剂进行封端,进一步提升了生产成本。
技术实现要素:
7.本发明提供一种氨基硅油的制备方法,其能够解决现有技术中氨基硅油制备工艺繁琐、反应周期长、原料成本高等技术问题。
8.本发明的技术方案是,一种氨基硅油的制备方法,包括以下步骤:
9.以二甲基硅氧烷混合物为起始原料,加入催化剂、水、硅烷偶联剂混合均匀,升温至50~90℃保温10~120min,然后进行真空脱低处理,再升温至95~145℃,反应1~4h,最后真空脱低处理,即得氨基硅油,整个制备过程均在氮气保护下进行。
10.进一步地,所述二甲基硅氧烷混合物为二甲水解物,其中含有线体和环体,环体比例占混合物的50~80%,优选60~75%。
11.进一步地,所述催化剂为四甲基氢氧化铵,氢氧化钾,氢氧化钠,氢氧化锂中的一种或几种。
12.进一步地,所述的硅烷偶联剂为γ-氨丙基甲基二乙氧基硅烷(fd-552),γ-氨丙基甲基二甲氧基硅烷(fd-554),n-β-(氨乙基)-γ-氨丙基甲基二甲氧基硅烷(fd-602),γ-二乙烯三氨基丙基甲基二甲氧基硅烷(fd-892),γ-哌嗪基丙基甲基二甲氧基硅烷(fd-1638)其中的一种或几种。
13.进一步地,二甲基硅氧烷混合物、催化剂、水和硅烷偶联剂的质量比为1000:0.01~0.85:0~8.5:10~85,优选比例为1000:0.01~0.5:0~8:10~80。
14.进一步优选地,水的加入量为硅烷偶联剂质量的0~9%。
15.进一步优选地,第一次升温时,温度控制为70~85℃,保温时间为25~30min;第二次升温时,温度为100~140℃,时间为2~3h。
16.进一步地,脱低处理时,体系的真空度在-0.06~-0.1mpa。
17.进一步地,整个制备过程均进行搅拌,搅拌速率为200r/min~800r/min。
18.进一步地,氮气通入速率为50~200ml/min。优选50~150ml/min。
19.采用上述方法,可以制备氨基硅油的结构式为:
[0020][0021]
其中,r2,r3分别为-ch3,-och3,-och2ch3,-oh中的一种或几种,r1的结构为-ch2ch2ch2nh2,-ch2ch2ch2nhch2ch2nh2,-ch2ch2ch2nhch2ch2ch2n(ch3)2,或者中的一种或几种;同时,r1结构中的氨基可以被伯胺,叔胺,仲胺,季铵盐和哌嗪所代替。
[0022]
本发明具有以下有益效果:
[0023]
1、二甲水解物中既含有线体也有环体,现有技术一般采用二甲基硅氧烷混合环体(dmc)或者用其精馏提纯后的八甲基环四硅氧烷(d4)为原料,需要对水解物进行环线分离,以分离后的环体dmc或者d4为原料制备氨基硅油。本发明直接以二甲水解物作为原料,省略了环线分离及纯化步骤,水解物原料获取渠道广,工艺简单,能够大幅降低成本,且整个反应过程仅需一次加料法,采用一锅法制备,操作简单,反应周期短,同时减少了中途加料操作可能带入外界空气中的氧气和水对反应产生的影响。
[0024]
2、本发明直接以二甲水解物为原料,通过控制线体和环体在一定的比例下,加入硅烷偶联剂、催化剂和水,先控制较低温度,此时环体不进行开环反应,但线体与硅烷偶联
剂进行反应生成甲氧基封端的氨基中间体,然后进行升温至环体开环的温度,环体进行反应,前期反应生成的甲氧基封端的氨基中间体可以作为封端剂对开环聚合起到链终止作用。另外线体所含有的羟基在催化剂作用下也会缩合脱出水分子,使得硅烷偶联剂水解更加充分。但过多的线体含量占比会导致反应过程中不断缩合产生水分,造成聚合反应正向平衡被抑制,从而影响环体的开环聚合,因此选择环体占比50~80%的水解物为原料,优选环体占比为60~70%。
[0025]
3、本发明中通过控制水的添加量,达到线体羟基与水中羟基总值平衡来控制硅烷偶联剂水解的程度,得到烷氧基封端、分子量高低不同具有对后期反应封端功能的低聚物氨基小分子,从而控制体系中封端体的属性及含量,无须额外添加封端剂,即可制备粘度可控(封端体含量)、储存稳定(无羟基属性的烷氧基封端产物),外观澄清透明的氨基硅油。
[0026]
4、本发明所涉及的合成工艺,在升温保温过程中,通过搅拌和n2鼓泡作用使得硅烷偶联剂与二甲水解物均匀混合。在有水和碱性的存在环境下,硅烷偶联剂在较短的时间里也能充分水解脱去醇类小分子,形成烷氧基封端产物。同时由于醇类小分子的沸点相对较低,在高温减压条件下容易从体系中脱离,不会对后续反应过程产生影响。
具体实施方式
[0027]
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。
[0028]
以下实施例中的各阶段产物性能按如下方法测量计算:
[0029]
1)收率计算:将最终得到的成品质量与实验初总投料量比值的百分数%。
[0030]
2)粘度测定:采用博勒飞标准粘度计dv2t测试。
[0031]
3)氨值测定:采用hg/t 4260-2011规定方法测定。
[0032]
4)挥发分测定:快速水分测定仪(仪器:梅特勒托利多hs153)。
[0033]
实施例1
[0034]
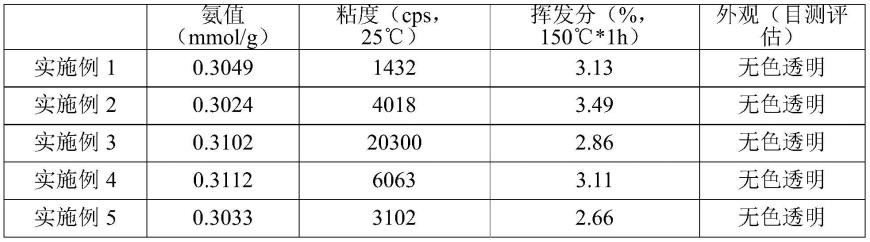
在安装有温度计,氮气导管,电动搅拌器和直形冷凝管的四颈烧瓶里,加入1000.00g二甲水解物(环线比为6.8:3.2),30.20g fd-602,0.35g四甲基氢氧化铵,1.31g水,常压下以50ml/min氮气保护,搅拌升温至85℃,保温30分钟,随后维持真空-0.08mpa半小时脱除低分子。升温并保持温度110℃反应3小时,随后升高温度至140℃维持1小时使催化剂失活,真空脱低后降温冷却得低沸物78.62g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油918.10g,收率为88.78%,氨值0.3049mmol/g,粘度1432cps,挥发分3.13%。
[0035]
实施例2
[0036]
在安装有温度计,氮气导管,电动搅拌器和直形冷凝管的四颈烧瓶里,加入1000.12g二甲水解物(环线比为6.8:3.2),30.03g fd-602,0.35g四甲基氢氧化铵,2.09g水,常压下以100ml/min氮气保护,搅拌升温至85℃,保温30分钟,随后维持真空-0.08mpa半小时脱除低分子。升温并保持温度110℃反应3小时,随后升高温度至140℃维持1小时使催化剂失活,真空脱低后降温冷却得低沸物76.79g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油916.32g,收率为88.74%,氨值0.3024mmol/g,粘度4018cps,挥发分3.49%。
[0037]
实施例3
[0038]
在安装有温度计,氮气导管,电动搅拌器和直形冷凝管的四颈烧瓶里,加入
1000.23g二甲水解物(环线比为6.8:3.2),30.15g fd-602,0.35g四甲基氢氧化铵,2.35g水,常压下以150ml/min氮气保护,搅拌升温至85℃,保温30分钟,随后维持真空-0.08mpa半小时脱除低分子。升温并保持温度110℃反应3小时,随后升高温度至140℃维持1小时使催化剂失活,真空脱低后降温冷却得低沸物83.85g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油915.82g,收率为88.65%,氨值0.3102mmol/g,粘度20300cps,挥发分2.86%。
[0039]
实施例4
[0040]
在实施例2相同工艺操作条件基础上,将二甲基硅烷混合环体的环线比调整为(6.0:4.0),其中二甲水解物1000.33g,30.42g fd-602,0.36g四甲基氢氧化铵,2.09g水。反应结束后得低沸物82.79g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油918.42g,收率为88.87%,氨值0.3112mmol/g,粘度6063cps,挥发分3.11%。
[0041]
实施例5
[0042]
在实施例2相同工艺操作条件基础上,将二甲基硅烷混合环体的环线比调整为(7.5:2.5),其中二甲水解物1000.30g,30.41g fd-602,0.35g四甲基氢氧化铵,2.09g水。反应结束后得低沸物83.28,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油915.27g,收率为88.57%,氨值0.3033mmol/g,粘度3102cps,挥发分2.66%。
[0043]
实施例6
[0044]
在实施例2的基础上,将催化剂改为0.10g的koh,反应条件为130℃保持2h,其它工艺保持不变,其中二甲水解物1000.31g,30.40g fd-602,0.35g四甲基氢氧化铵,2.09g水。反应结束后得低沸物79.15g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油919.83g,收率为88.89%,氨值0.3038mmol/g,粘度4130cps,挥发分3.21%。
[0045]
实施例7
[0046]
在实施例2的基础上,将催化剂改为0.2g的lioh,反应条件为135℃保持2h,其它工艺基本保持不变,其中二甲水解物1000.28g,30.12g fd-602,2.09g水。反应结束后得低沸物80.75g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油912.27g,收率为88.34%,氨值0.3038mmol/g,粘度3982cps,挥发分3.62%。
[0047]
实施例8
[0048]
在实施例2的基础上,将偶联剂替换成80.03gfd-552,其它工艺保持不变,其中二甲水解物1000.12g,2.09g水,0.35g四甲基氢氧化铵。反应结束后得低沸物81.42g,主产为氨丙基结构无色透明粘稠的氨基硅油950.83g,收率为87.82%,氨值0.4642mmol/g,粘度146cps,挥发分4.11%。
[0049]
实施例9
[0050]
在实施例2的基础上,将偶联剂替换成10.21gfd-554,其它工艺保持不变,其中二甲水解物1000.22g,2.09g水,0.34g四甲基氢氧化铵。反应结束后得低沸物75.36g,主产为氨丙基结构无色透明粘稠的氨基硅油902.56g,主产为氨丙基结构无色透明粘稠的氨基硅油,收率为89.11%,氨值0.0511mmol/g,粘度12630cps,挥发分3.26%。
[0051]
实施例10
[0052]
在实施例2的基础上,将偶联剂替换成13.90gfd-552和11.85gfd-554的混合物,其它工艺保持不变,其中二甲水解物1000.52g,2.09g水,0.35g四甲基氢氧化铵。反应结束后得低沸物78.59g,主产为氨丙基结构无色透明粘稠的氨基硅油912.67g,主产为氨丙基结构
无色透明粘稠的氨基硅油,收率为88.72%,氨值0.1531mmol/g,粘度4130cps,挥发分3.21%。
[0053]
实施例11
[0054]
在实施例2的基础上,将偶联剂替换成36.4gfd-892,其它工艺保持不变,其中二甲水解物1000.02g,2.09g水,0.35g四甲基氢氧化铵。反应结束后得低沸物81.23g,主产为氨丙基结构无色透明粘稠的氨基硅油918.46g,主产为二乙烯基三氨基结构无色透明粘稠的氨基硅油,收率为88.41%,氨值0.4321mmol/g,粘度4100cps,挥发分3.55%。
[0055]
实施例12
[0056]
在实施例2的基础上,将偶联剂替换成33.82gfd-1638,其它工艺保持不变,其中二甲水解物1000.32g,2.09g水,0.34g四甲基氢氧化铵。反应结束后得低沸物81.06g,主产为氨丙基结构无色透明粘稠的氨基硅油916.43g,主产为哌嗪基结构无色透明粘稠的氨基硅油,收率为88.41%,氨值0.2918mmol/g,粘度4261cps,挥发分3.46%。
[0057]
对比例1
[0058]
在实施例3的基础上,将水的用量调整至2.88g,其它工艺保持不变。其中1000.24g二甲水解物(环线比为6.8:3.2),30.17g fd-602,0.35g四甲基氢氧化铵,反应结束后得低沸物110.18,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油876.84g,收率为84.83%,氨值0.3326mmol/g,粘度31800cps,挥发分3.96%。
[0059]
对比例2
[0060]
在实施例1的基础上,将二甲基硅烷混合环体的环线比调整为(4:6),其它工艺保持不变,其中1000.02g二甲水解物,30.20g fd-602,0.35g四甲基氢氧化铵,1.31g水,反应结束后得低沸物93.69g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油909.50g,收率为88.14%,氨值0.3046mmol/g,粘度42600cps,挥发分2.51%。
[0061]
对比例3
[0062]
在实施例2的基础上,将二甲水解物的环线比调整为(4:6),其它工艺保持不变,其中1000.12g二甲水解物,30.02g fd-602,0.35g四甲基氢氧化铵,2.09g水,反应结束后得低沸物86.11g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油915.69g,收率为88.68%,氨值0.3074mmol/g,粘度10032cps,挥发分2.88%。
[0063]
对比例4
[0064]
在实施例2的基础上,将二甲水解物的环线比调整为(9:1),其它工艺保持不变,其中1000.12g二甲水解物,30.01g fd-602,0.36g四甲基氢氧化铵,2.10g水,反应结束后得低沸物96.44g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油904.86g,收率为87.63%,氨值0.3053mmol/g,粘度1300cps,挥发分2.64%。
[0065]
对比例5
[0066]
在对比例4的基础上,将水用量调整为2.70g,其它工艺保持不变,其中1000.12g二甲水解物(环线比9:1),30.01g fd-602,0.36g四甲基氢氧化铵,反应结束后得低沸物107.66g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油893.92g,收率为86.52%,氨值0.3112mmol/g,粘度4300cps,挥发分2.99%。
[0067]
对比例6
[0068]
在实施例3的基础上,将氮气流速调整至10ml/min,其它工艺保持不变,其中
1000.25g二甲水解物(环线比为6.8:3.2),30.17g fd-602,0.35g四甲基氢氧化铵,2.35g水.反应结束后得低沸物110.13g,主产为氨乙基氨丙基结构无色透明粘稠的氨基硅油891.89g,收率为86.33%,氨值0.3122mmol/g,粘度14030cps,挥发分2.78%。
[0069]
以上实施例反应过程中,存在部分小分子被抽入真空系统从而产生损失影响氨基硅油收率。
[0070]
对上述实施例所得到的氨基硅油与市面上购买的氨基硅油(c803(星火有机硅),6409(科士康))进行性能测试对比,主要测试内容包括其粘度、挥发分、氨值、储存稳定性以及应用于棉布后的手感和柔软度评估。
[0071]
表1实施例1~12、对比例所得到氨基硅油与市售氨基硅油的一般性能指标统计如下:
[0072][0073][0074]
由实施例1、2、3性能指标可以看出,在其它配方比例不变的情况下,调整水的用量,可以明显控制氨基硅油产品的粘度。
[0075]
由实施例2、4、5性能指标可以看出,当环线比改变时,硅油的粘度也随之发生变化,这是由于二甲水解物中的线体能在反应过程中通过缩合提供一部分水分促进硅烷偶联剂的水解。
[0076]
由实施例2和实施例6-12的性能指标对比可以看出,在仅改变偶联剂种类不改变
其物质的量的情况下,最终得到氨基硅油粘度相对稳定。当仅改变硅烷偶联剂的用量时,其粘度随着硅烷偶联剂用量的增加而减小,因为封端剂的增加降低了原油的分子量;其转化率随着硅烷偶联剂用量的增加而降低,当偶联剂增加时,抑制平衡反应正向进行,使收率降低。
[0077]
当二甲水解物中环线比过低时,如对比例2、3,在添加相同的水的情况下,与实施例1、2相比粘度会上涨非常明显,这是由于体系中线体含量高,本身缩合产生大量水,使得体系中存在大量羟基,羟基间互相缩合使得粘度持续上涨。
[0078]
当二甲水解物中环线比过高时,由于缺少线体提供的水,硅烷偶联剂水解不充分,最终原油更多以甲氧基进行封端,得到的原油粘度低。当需要得到较高粘度原油时,需要加入大量的水进行水解,过多的水同时会影响聚合反应的正向进行,使得体系聚合度下降。如对比例4、5,在添加相同的水的情况下,与实施例2、3相比粘度均发生下降,同时收率都明显偏低。
[0079]
对比例1相较于实施例3采用了过量的水,其粘度得到了明显提升,这是由于硅烷偶联剂水解更彻底,甲氧基全部被水解形成硅羟基,体系中的羟基发生缩合使得封端剂减少,粘度上涨。同时缩合产生的水会抑制环体开环的正向进行,使得原料的收率降低。
[0080]
对比例6相较于实施例3用了较低的氮气流量,其粘度明显下降,同时收率降低,这是由于过低的氮气速率使得反应过程体系中产生的甲醇没有被完全带出,在聚合过程中参与封端,导致收率降低,体系粘度减小。
[0081]
验证例1:氨基硅油储存稳定性测试
[0082][0083]
硅油粘度随着时间变化越小,其储存稳定越好。由表中可知,由环线比较低的二甲基硅烷混合物制备的氨基硅油(对比例2、对比例3)和使用过量水制备的氨基硅油(对比例1)储存稳定性较差,这是因为体系中含有大量羟基封端的分子链存在,在储存过程中发生缩合使得原油粘度进一步增大。
[0084]
验证例2:氨基硅油应用性能测试
[0085]
分别取实施例1-3制备的氨基硅油和市售氨基硅油,在酸性水溶液和乳化剂的作用下乳化成固含量在27%的透明乳液。随后取1g乳化好的硅油乳液,按1:100的比例稀释成整理织物所用的工作浴液。取面积为10cm x 10cm白色纯棉胚布(规格为21x21x108x98),在浴液中充分浸泡后,用小型变频轧车二浸二轧进行处理,带液率为60%,随后用将织物在定型烘干小样机中用140℃烘干90s,室温平衡24h后进行性能测试,柔软度以弯曲刚度表示,
测试通过电脑柔软度仪进行测定。
[0086]
表2氨基硅油整理后棉织物的应用性能
[0087][0088][0089]
弯曲刚度越小,代表柔软性越好。实验验证结果表明,本发明所制备的氨基硅油具有良好的柔软整理性能。
[0090]
验证例3:织物手感评估
[0091]
织物手感是一种个人主观评判的结果,此次实验我们将织物手感分为软和滑两方面进行评估,邀请7人盲摸打分的方式,将手感分为1到5级,最好为5级,最差为1级,最后综合七人评分的平均值,对整理后的织物的手感进行排序。
[0092]
表3织物手感评估
[0093][0094]
从表3结果可知,经过处理后的织物手感无论从软还是滑方面都明显优于空白样,同时实施例1~7整理效果均不差于两款市售氨基硅油,其中实施例1较低粘度的氨基硅油对织物的整理效果最佳。
[0095]
由此可见,由本合成方法一锅法制备的氨基硅油不仅具有方法简单,粘度可控等
优点,其使用性能相对于市场同类产品具有一定优势。
[0096]
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本技术中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1