一种基于rac-2-Br-DMNPA的蛋白质固相化学连接方法
一种基于rac-2-br-dmnpa的蛋白质固相化学连接方法
技术领域
1.本发明涉及多肽和/或蛋白质合成技术领域,具体涉及一种基于rac-2-br-dmnpa的蛋白质固相化学连接方法
背景技术:
2.1963年merrifield引入的固相多肽合成(spps)技术极大地促进了多肽和蛋白质的合成(r.b.merrifield,j.am.chem.soc.,1963,85,14,2149
–
2154.)。spps策略简化了多肽合成过程中的纯化步骤,可通过添加过量的试剂来保证化学反应彻底进行,并且适用于多肽的自动化合成。但随着合成多肽序列长度的增加,在氨基酸数量达到50个以上时,通过spps获得完整肽段就会变得困难,而天然化学连接(ncl)的出现为在水相中合成多肽和蛋白质提供了新的策略。ncl通过可逆的硫酯化转移和不可逆的s-n酰基转移反应,在两个连接肽段的n端和c端之间形成一个天然的肽键,连接无保护的多肽片段来得到全长蛋白,具有高度的化学选择性(t.wieland,liebigs ann.chem.,1953,583,129-149.)。
3.为了获得大蛋白,更有效的多段ncl连接策略(c.zuo,org.biomol.chem.,2019,17,727-744.)和用于一锅法的新型巯基保护基团及其脱保护策略(s.k.maity,angew.chem.int.ed.,2016,55,8108-8112.)逐步得到发展。但是,大型蛋白质的合成通常需要几个肽段的顺序组装,导致合成期间需要繁杂的纯化分离步骤,并且面对部分难溶的多肽或蛋白时,处理和纯化往往变得十分困难。
4.此外,一些巯基保护基团在spps时的早期引入是强制性的,这限制了它们在蛋白质合成中的广泛适用性。为了克服这个问题,利用对光不稳定的2-硝基苯衍生物作为自由半胱氨酸巯基的后期按需保护的策略得到发展,但对其蛋白质化学合成中的应用还缺乏更深入的研究(r.k.mcginty,nature,2008,453,812-816.)。作为2-硝基苯的衍生物,rac-2-br-dmnpa是一种新型的双功能巯基后期保护基。在紫外光的照射下,可以实现巯基保护的快速脱除。采用rac-2-br-dmnpa作为巯基后期的正交保护基,能实现蛋白的一锅法连接、脱硫及脱acm。但是在蛋白合成期间,仍难以避免多肽溶解性产生的不利影响以及脱保护后反应试剂的积累导致的液相纯化分离困难等问题。
5.因此,有必要开发一种合成简单、纯化便捷、产率可观、适用于半胱氨酸后期正交保护、能快速切割脱除的蛋白质固相连接方法,用以解决水溶性较差、难以分离的蛋白质的化学合成问题。
技术实现要素:
6.为解决相关问题,本发明的首要目的在于提供一种基于rac-2-br-dmnpa的蛋白质固相化学连接方法。
7.本发明的另一目的在于提供上述基于rac-2-br-dmnpa的蛋白质固相化学连接方法在多肽/蛋白质合成中的应用。
8.为了实现上述发明目的,本发明采用以下技术方案:
9.一种基于rac-2-br-dmnpa的蛋白质固相化学连接方法,包括如下步骤:
10.(1)spcl树脂的制备:
11.采用fmoc固相多肽合成法,以am pega树脂为载体,从c端到n端依次缩合n个fmoc-ala-cooh,fmoc-(β)ala-oh,以及rac-2-br-dmnpa,洗涤,干燥,获得am pega-nala-(β)ala-rac-2-br-dmnpa树脂,简称spcl(solid phase chemical ligation)树脂;n为1-4之间的整数;
12.(2)根据目的多肽或蛋白的序列,从任意两个半胱氨酸划分为片段1、片段2、片段3,确保片段2和片段3的n端为半胱氨酸,同时片段2的中间段有至少一个半胱氨酸;
13.(3)片段1、片段2、片段3的线性多肽树脂的制备:
14.使用c端为酰肼(-nhnh2)末端的树脂,简称酰肼树脂,采用fmoc固相多肽合成法,分别根据片段1、片段2、片段3的序列,从c端到n端依次缩合fmoc保护氨基酸,洗涤,干燥,获得片段1、片段2、片段3的线性多肽树脂;其中,
15.片段2的n端的半胱氨酸侧链的巯基采用噻唑烷(thz)作为保护基,其余半胱氨酸侧链的巯基采用乙酰胺甲基(acm)或三苯甲基(trt)作为保护基;
16.(4)c端为酰肼的多肽片段2、多肽片段3的制备:
17.取步骤(3)制备得到的片段2、片段3的线性多肽树脂,加入切割试剂,使多肽链从酰肼树脂上脱下,并脱除剩余侧链保护基;经过滤、旋干、萃取、离心、冻干,得到c端为酰肼的片段2、片段3粗品;进一步分离纯化,冻干,得到c端为酰肼的多肽片段2、多肽片段3;
18.(5)c端为mpaa的多肽片段1的制备:
19.取步骤(3)制备得到的片段1的线性多肽树脂,加入切割试剂,使多肽链从酰肼树脂上脱下,并脱除剩余侧链保护基;经过滤、旋干、萃取、离心、冻干,得到c端为酰肼的片段1粗品;溶解,加入乙酰丙酮,摇匀,再加入mpaa(4-巯基苯基乙酸),第一步反应,加入tcep(三(2-羧乙基)膦),第二步反应,离心,过滤,进一步分离纯化,冻干,得到c端为mpaa的多肽片段1;
20.(6)负载多肽片段2的spcl树脂的制备:
21.取步骤(4)制备得到的c端为酰肼的片段2,溶解,加入步骤(1)制备得到的spcl树脂,第一步反应,洗涤,重复第一步反应,洗涤,加入2-巯基乙醇,第三步反应,洗涤,得到负载多肽片段2的spcl树脂;
22.(7)负载c端为mpaa的多肽片段2的spcl树脂的制备:
23.乙酰丙酮和mpaa溶于缓冲盐溶液,与步骤(6)制备得到的负载多肽片段2的spcl树脂混合,反应,洗涤,得到负载c端为mpaa的多肽片段2的spcl树脂;
24.(8)负载连接肽1的spcl树脂的制备:
25.取步骤(4)制备得到的多肽片段3,溶解,和步骤(7)制备得到的负载c端为mpaa的多肽片段2的spcl树脂混合,进行自然化学连接(native chemical ligation,简称ncl)反应,洗涤,得到负载连接肽1的spcl树脂;
26.(9)负载n端为游离半胱氨酸的连接肽1的spcl树脂的制备:
27.哌啶溶于缓冲盐溶液,得混合液1,取步骤(8)制备得到的负载连接肽1的spcl树脂,加入混合液1,第一步反应,洗涤;甲氧胺盐酸盐、tcep、盐酸胍溶于缓冲盐溶液,得混合液2,加入上述反应体系,第二步反应,洗涤,得到负载n端为游离半胱氨酸的连接肽1的spcl
树脂;
28.(10)负载连接肽2的spcl树脂的制备:
29.取步骤(5)制备得到的c端为mpaa的片段1,溶解,和步骤(9)制备得到的负载n端为游离半胱氨酸的连接肽1的spcl树脂混合,进行自然化学连接反应,洗涤,得到负载连接肽2的spcl树脂;
30.(11)负载连接肽2的spcl树脂的紫外光解:
31.取步骤(10)制备得到的负载连接肽2的spcl树脂,加入预先配置的光解溶液,通过紫外光光照,使连接肽2从spcl树脂上脱下,离心,过滤,进一步分离纯化,冻干,得到目的多肽或蛋白。
32.所述的方法中,目的多肽或蛋白的序列随机,长度优选在1000个氨基酸以内,更优选在500个氨基酸以内,最优选在200个氨基酸以内。
33.所述的酰肼树脂优选为2-chlorotrityl chloride树脂(简称2-cl树脂)经5%(v/v)水合肼/dmf处理30min后得到获得的酰肼树脂。
34.所述的fmoc保护氨基酸与am pega树脂的偶联时间优选为4~6h;更优选为4h。
35.所述的fmoc保护氨基酸与酰肼树脂的偶联时间优选为2~4h;更优选为2h。
36.步骤(1)与步骤(3)中所述的从c端到n端依次缩合fmoc保护氨基酸的具体操作为:在偶联体系作用下先将第1位氨基酸和am pega或脱除fmoc后的酰肼树脂反应生成氨基酸-氨基树脂,再逐一偶联其它fmoc保护氨基酸,获得线性多肽树脂。
37.所述的am pega树脂偶联体系中的氨基酸缩合剂优选为“tbtu+dipea”。
38.所述的am pega树脂偶联体系中的rac-2-br-dmnpa缩合剂优选为“dcc”。
39.所述的rac-2-br-dmnpa的用量,优选按5倍当量的am pega树脂装载量计算。
40.所述的酰肼树脂偶联体系中的氨基酸缩合剂优选为“hobt+dic”或“tbtu+diea”。
41.所述的偶联体系中的fmoc脱保护试剂优选为20%哌啶/dmf。
42.步骤(4)与步骤(5)中所述的切割的试剂优选为三氟乙酸(tfa)、水和1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(dodt)按95:2.5:2.5的体积比混合得到的溶液。
43.步骤(4)与步骤(5)中所述的切割的时间优选为2~4h。
44.步骤(4)与步骤(5)中所述的萃取的萃取剂优选为冰乙醚。
45.步骤(4)与步骤(5)中所述的萃取的次数优选为两次。
46.步骤(4)、与步骤(5)中所述的分离纯化优选通过反向液相色谱实现。
47.所述的反向液相色谱的流动相为含0.1%三氟乙酸的乙腈/水混合液。
48.步骤(5)中溶解片段1粗品的溶液优选为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),溶液的ph值为3.0
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),溶液的ph值为3.0。
49.步骤(5)中所述的乙酰丙酮的用量,优选按片段1粗品:乙酰丙酮=1:2.5的摩尔比计算,所述的摇匀时间优选为3分钟。
50.步骤(5)中所述的mpaa的用量,优选按片段1粗品:mpaa=1:10的摩尔比计算。
51.步骤(5)中所述的第一步反应的条件为温度20~30℃(室温),反应时间优选为12小时。
52.步骤(5)中所述的tcep的用量,优选按片段1粗品:tcep=1:10的摩尔比计算。
53.步骤(5)中所述的第二步反应的条件为温度20~30℃(室温),反应时间优选为20分钟。
54.步骤(6)中溶解片段2的溶液优选为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),溶液的ph值为3.0
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),溶液的ph值为7.5。
55.步骤(6)中所述的片段2的用量优选按1倍当量的spcl树脂装载量计算。
56.步骤(6)中所述的第一步反应的条件为温度20~30℃(室温),反应时间优选为4小时。
57.步骤(6)中所述的2-巯基乙醇的用量,优选按片段2:2-巯基乙醇=1:20的摩尔比计算。
58.步骤(6)中所述的第三步反应的条件为温度20~30℃(室温),反应时间优选为60分钟。
59.步骤(7)中溶解乙酰丙酮和mpaa的溶液优选为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),溶液的ph值为3.0
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),溶液的ph值为3.0。
60.步骤(7)中所述的乙酰丙酮的用量,优选按片段2:乙酰丙酮=1:2.5的摩尔比计算,所述的摇匀时间优选为3分钟。
61.步骤(7)中所述的mpaa的用量,优选按片段2:mpaa=1:40的摩尔比计算。
62.步骤(7)中所述的反应的条件为温度20~30℃(室温),反应时间优选为6小时。
63.步骤(7)中所述的tcep的用量,优选按片段2:tcep=1:10的摩尔比计算。
64.步骤(7)中所述的第二步反应的条件为温度20~30℃(室温),反应时间优选为20分钟。
65.步骤(8)中溶解片段3的溶液为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),0.15~0.25mol/l mpaa,38~42mmol/l tcep,溶液的ph值为6.5
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),0.2mol/l mpaa,40mmol/l tcep,溶液的ph值为6.5。
66.步骤(8)中所述的片段3的用量优选按1.3倍当量的spcl树脂装载量计算。
67.步骤(8)中所述的自然化学连接反应的条件为温度37
±
0.5℃,反应时间优选为8~12小时。
68.步骤(9)中所述的混合液1优选为20%(v/v)哌啶,5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),溶液的ph值为3.0
±
0.5;进一步优选为20%(v/v)哌啶,6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),溶液的ph值为3.0。
69.步骤(9)中所述第一步反应的条件为温度20~30℃(室温),反应溶液ph优选为11.0,反应时间优选为20-40分钟。
70.步骤(9)中所述的混合液2优选为0.15~0.25mol/l甲氧胺盐酸盐、9~11mm tcep、5.5~6.5mol/l盐酸胍、0.15~0.25mol/l pbs、溶液的ph值为4.0
±
0.5;进一步优选为0.2mol/l甲氧胺盐酸盐、10mm tcep、6mol/l盐酸胍、0.2mol/l pbs、溶液的ph值为4.0。
71.步骤(9)中所述第二步反应的条件为温度20~30℃(室温),反应溶液ph优选为4,反应时间优选为2-4小时;
72.步骤(10)中所述的溶解片段1的溶液为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),0.15~0.25mol/l mpaa,38~42mmol/l tcep,溶液的ph值为6.5
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),0.2mol/l mpaa,40mmol/l tcep,溶液的ph值为6.5。
73.步骤(10)中所述的片段1的用量优选按1倍当量的spcl树脂装载量计算。
74.步骤(10)中所述的自然化学连接反应的条件为温度37
±
0.5℃,反应时间优选为8~12小时。
75.步骤(11)中所述的光解溶液优选为5.5~6.5mol/l盐酸胍,0.15~0.25mol/l磷酸二氢钠(pbs),9~11mm tcep,350~450mm盐酸氨基脲(sem),溶液的ph值为3.0
±
0.5;进一步优选为6mol/l盐酸胍,0.2mol/l磷酸二氢钠(pbs),10mm tcep,400mm盐酸氨基脲,溶液的ph值为3.0。
76.步骤(11)中所述的紫外光光照的条件为温度4℃,紫外光波长为365nm,时间优选为30~90分钟。
77.步骤(11)中所述的分离纯化优选通过反向液相色谱实现。
78.所述的反向液相色谱的流动相为含0.1%三氟乙酸的乙腈/水混合液。
79.上述基于rac-2-br-dmnpa的蛋白质固相化学连接方法在蛋白质化学合成中的应用。
80.本发明方法的原理如下:采用fmoc固相多肽合成法,在am pega树脂上依次缩合fmoc-ala-cooh(1-4个)、fmoc-(β)ala-cooh(1个),并以此短肽为连接子与rac-2-br-dmnpa缩合,得到能与多肽侧链的巯基选择性结合/脱除的spcl树脂。设计三段氨基酸序列相近的多肽片段,以酰肼树脂为载体,从c端到n端依次缩合fmoc保护氨基酸,得到相应的酰肼树脂;其中,多肽片段2使用fmoc-cys(thz)-oh作为其n端半胱氨酸的保护基团(第1位),其中第5位的半胱氨酸巯基为片段2与spcl树脂的结合位点;多肽片段3的n端为半胱氨酸(第1位);多肽片段2的裸露巯基与rac-2-br-dmnpa反应,从而被spcl树脂固定,经过固相硫酯转化,得到负载c端为mpaa的多肽片段2的spcl树脂;然后与n端为半胱氨酸的多肽片段3进行第一步ncl反应,得到负载连接肽1的spcl树脂;随后依次进行固相脱fmoc与脱thz反应,使连接肽1的n端半胱氨酸得以暴露;接着与c端为mpaa的多肽片段1进行第二步ncl反应,得到负载连接肽2的spcl树脂;最后通过紫外光照反应将连接肽2从树脂上裂解下来并纯化,即可得到连接肽2。
81.本发明相对于现有技术具有如下的优点及效果:
82.本发明方法提供了在am pega树脂上引入rac-2-br-dmnpa并进行全程固相化学连接合成多肽实例,仅需要最后一步的液相色谱纯化即可得到目标产物,大大简化了繁琐的合成多肽/蛋白质的纯化分离步骤。
83.本发明方法中为rac-2-br-dmnpa与am pega树脂设计了不同长度的中间连接子(序列长度为2-5个氨基酸的短肽),可根据实际合成多肽/蛋白质的序列长度、树脂固定化位点、空间位阻等因素,选择长度较为合适的中间连接子以优化固相化学连接反应。
84.本发明方法中使用rac-2-br-dmnpa作为双功能标签,在spcl树脂上实现了按需的多肽后期巯基保护,同时实现巯基保护的快速紫外光解脱除。
85.本发明方法中使用spcl树脂成功实现了n-c及c-n的多肽固相化学连接反应;经
spcl树脂固定化后的多肽片段2可在固相上直接进行多肽酰肼-硫酯转化反应,并具有90%以上的转化率;再者,spcl树脂固定化的多肽片段2在与片段3在ncl连接反应后,可依次进行固相脱fmoc与脱thz反应,均具有90%以上的转化率。
86.本发明方法能用于处理水溶性差的多肽,为难溶的多肽/蛋白质提供了一种进行固相反应的途径。
87.本发明方法将蛋白质合成中的纯化步骤简化为简单的洗涤步骤,在spcl树脂上进行任意反应后,通过固相洗涤即可实现反应混合物的分离,无需额外的液相纯化分离,也为最终产物的液相纯化分离提供了便利。
88.本发明方法中在获得c端酰肼的多肽片段1粗品后,通过反应体系ph 3的硫酯化反应,将c端酰肼转化为mpaa并分离、纯化、冻干,使mpaa末端的多肽片段1以冻干形式稳定存在,避免了mpaa在ph>7时容易发生的水解反应。
附图说明
89.图1为实施例1制得的多肽片段1的hplc示意图(a)和esi-ms表征图(b)。
90.图2为实施例1制得的多肽片段2的hplc示意图(a)和esi-ms表征图(b)。
91.图3为实施例1制得的多肽片段3的hplc示意图(a)和esi-ms表征图(b)。
92.图4为实施例1制得的负载多肽片段2的spcl树脂的紫外光照切割反应随时间变化的hplc示意图(a)和esi-ms表征图(b)。
93.图5为实施例1制得的负载c端为mpaa的多肽片段2的spcl树脂的紫外光照切割反应随时间变化的hplc示意图(a)和esi-ms表征图(b)。
94.图6为实施例1制得的负载连接肽1的spcl树脂的紫外光照切割产物的hplc示意图(a)和esi-ms表征图(b)。
95.图7为实施例1制得的负载连接肽2的spcl树脂的紫外光照切割产物的hplc示意图(a)和esi-ms表征图(b)。
具体实施方式
96.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
97.下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
98.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
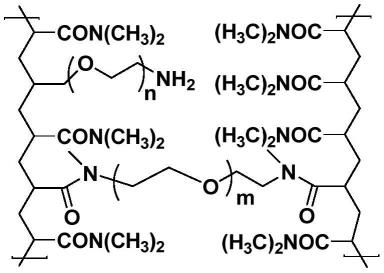
99.下述实施例中使用的am pega树脂的结构式如下:
100.101.下述实施例中使用的rac-2-br-dmnpa的结构式为:
[0102][0103]
其余fmoc保护氨基酸为fmoc-cys(thz)-oh、fmoc-cys(trt)-oh、fmoc-ala-oh、fmoc-(β)ala-oh、fmoc-leu-oh、fmoc-lys(boc)-oh、fmoc-gly-oh。
[0104]
下述实施例中hplc,其仪器为安捷伦1260,色谱柱为phenomenex c18柱,流动相为水和乙腈(含0.1%(v/v)tfa)。
[0105]
下述实施例中合成的三种多肽片段序列为如下,而且合成的多肽c端均为酰胺化:
[0106]
多肽片段1:alkcglkg;
[0107]
多肽片段2:c(thz)alkcglkg;
[0108]
多肽片段3:calkaglkg。
[0109]
下述实施例中用到的试剂名称及缩写:
[0110]
dmf:n,n-二甲基甲酰胺;
[0111]
dcm:二氯甲烷;
[0112]
hobt:1-羟基苯并三唑;
[0113]
dic:n,n-二异丙基碳二亚胺;
[0114]
tbtu:苯并三唑四甲基四氟硼酸;
[0115]
diea:n,n-二异丙基乙胺;
[0116]
nmm:n-甲基吗啉;
[0117]
dcc:n,n'-二环己基碳二亚胺;
[0118]
dipea:n,n-二异丙基乙胺
[0119]
tfa:三氟乙酸;
[0120]
dodt:1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸;
[0121]
acn:乙腈;
[0122]
sem:盐酸氨基脲
[0123]
tcep:三(2-羧乙基)膦;
[0124]
mpaa:4-巯基苯基乙酸;
[0125]
pbs:磷酸二氢钠;
[0126]
ipa:异丙醇。
[0127]
实施例1:基于rac-2-br-dmnpa的三段多肽固相化学连接方法:
[0128]
(1)fmoc-ala-am pega树脂的制备:取1g am pega树脂于多肽合成管中,负载量为0.5mmol/g,加入15ml dmf和200μl diepa室温震荡溶胀30min,依次用dmf、dcm、dmf各洗涤两次,称取fmoc-ala-oh(1mmol)、tbtu(0.98mmol)以及dipea(2mmol),将fmoc-ala-oh与tbtu用少量dmf溶解,加入diepa,室温震荡反应活化羧基3min,之后将活化后的氨基酸加入到树脂中,在室温下震荡反应4h,依次用dmf、dcm、dmf各洗涤两次,往树脂里加入10ml乙酸酐/diepa/dmf(1/2/2,v/v/v),室温下震荡反应30min,封闭未反应的氨基,然后采用dmf、
dcm各洗涤三次,氮气吹干得到干燥树脂,取树脂通过紫外检测其树脂负载量,最终得到负载量约为0.44mmol/g的fmoc-ala-am pega树脂。
[0129]
(2)am pega-ala-ala-(β)ala-rac-2-br-dmnpa树脂(spcl树脂)的制备:取得到的fmoc-ala-am pega树脂加入15ml dmf室温震荡溶胀两次,每次15min,排干,往树脂加入10ml 20%哌啶/dmf,室温下震荡反应5min,再用dmf洗涤两次后,再次加入10ml 20%哌啶/dmf,室温下震荡反应5min,依次用dmf、dcm、dmf各洗涤两次,排干溶剂,得到脱除fmoc保护的树脂,称取fmoc-ala-oh(4
×
0.49mmol),tbtu(3.9
×
0.49mmol)以及diea(8
×
0.49mmol),将fmoc氨基酸与tbtu用少量dmf溶解,加入diea,室温震荡反应活化羧基3min,之后将活化后的氨基酸加入到树脂中,在室温下震荡反应2h,可用茚三酮试剂监测反应,用dmf、dcm、dmf各洗涤两次,重复以上实验操作(氨基酸更换为fmoc-(β)ala-oh),随后往树脂加入10ml 20%哌啶/dmf,室温下震荡反应5min,再用dmf洗涤两次后,再加入10ml 20%哌啶/dmf,室温下震荡反应5min,依次用dmf、dcm、dmf各洗涤两次,称取rac-2-br-dmnpa(5
×
0.49mmol)和dcc(5
×
0.49mmol),在避光氮气保护的条件下震荡12h,使用dmf、dcm各洗涤三次,ipa洗涤两次,乙醚洗涤两次,dcm洗涤4次,最后使用氮气吹干得到干燥树脂,得到am pega-ala-ala-(β)ala-rac-2-br-dmnpa树脂(spcl树脂),于4℃下避光保存。
[0130]
(3)fmoc-gly-nhnh2树脂的制备:取1g 2-chlorotrityl chloride树脂(简称2-cl树脂)于多肽合成管中,负载量为0.3-0.5mmol/g,加入15ml dmf室温震荡溶胀两次,每次30min,排干,往树脂加入15ml 5%水合肼/dmf,在室温下震荡反应30min,再用dmf洗涤两次后,第二次加入15ml 5%水合肼/dmf,室温下震荡反应30min,再用dmf洗涤两次后,第三次加入15ml 5%水合肼/dmf,室温下震荡反应30min,依次用dmf、dcm、dmf各洗涤两次,排干溶剂,得到c端为酰肼末端的树脂(简称酰肼树脂),往树脂加入15ml 5%meoh/dmf,室温下震荡反应10min,依次用dmf、dcm、dmf各洗涤两次,排干溶剂,封闭未被酰肼化的树脂位点,称取fmoc-gly-oh(1mmol)、tbtu(0.98mmol)以及diea(2mmol),将fmoc-gly-oh与tbtu用少量dmf溶解,加入diea,室温震荡反应活化羧基3min,之后将活化后的氨基酸加入到树脂中,在室温下震荡反应2h,采用dmf、dcm各洗涤三次,氮气吹干得到干燥树脂,取树脂通过紫外检测其树脂负载量,最终得到约负载量为0.51mmol/g的fmoc-gly-nhnh2树脂。
[0131]
(4)多肽片段1-nhnh2树脂、多肽片段2-nhnh2树脂、多肽片段3-nhnh2树脂的制备:取得到的fmoc-gly-nhnh2树脂加入15ml dmf室温震荡溶胀两次,每次15min,排干,之后往树脂里加入10ml 20%乙酸酐/dmf,室温震荡反应20min从而封闭未耦合上氨基酸的树脂的氨基,阻止其下一步反应,依次用dmf、dcm、dmf各洗涤两次,往树脂加入10ml20%哌啶/dmf,室温下震荡反应5min,再用dmf洗涤两次后,再次加入10ml 20%哌啶/dmf,室温下震荡反应5min,依次用dmf、dcm、dmf各洗涤两次,排干溶剂,得到脱除fmoc保护的树脂,称取fmoc-lys-oh(4
×
0.42mmol),tbtu(3.9
×
0.42mmol)以及diea(8
×
0.42mmol),将fmoc氨基酸与tbtu用少量dmf溶解,加入diea,室温震荡反应活化羧基3min,之后将活化后的氨基酸加入到树脂中,在室温下震荡反应2h,可用茚三酮试剂监测反应,用dmf、dcm、dmf各洗涤两次,重复以上实验操作(氨基酸用量以树脂的4倍摩尔当量计算,震荡反应为2h),按照多肽片段1序列从c端到n端进行耦合。合成完毕后采用dmf、dcm各洗涤三次,氮气吹干得到干燥树脂;多肽片段2-nhnh2树脂、多肽片段3-nhnh2树脂的制备方法与多肽片段1-nhnh2树脂的制备方法相同。
[0132]
(5)多肽片段1-nhnh2粗品的制备:取200mg步骤(4)得到的树脂,加入10ml切割试剂(tfa:dodt:h2o=体积比95:2.5:2.5),震荡反应2~4h,过滤得到黄色透明液体,液体旋蒸仪旋干后,加入约20ml冰乙醚萃取两次,离心后收集沉淀,将样品冻干,得到约45mg多肽片段1-nhnh2粗品。
[0133]
(6)多肽片段1-mpaa纯品的制备:取步骤(5)获得的多肽片段1-nhnh2粗品,用含6mol/l盐酸胍、0.2mol/l pbs、ph 3.0的缓冲盐溶液溶解,加入2.5倍多肽粗品当量的乙酰丙酮,震荡3分钟摇匀,再加入10倍多肽粗品当量的mpaa,反应12h,再加入10倍多肽粗品当量的tcep,反应20分钟,离心,过滤,通过流动相为含0.1%tfa的acn/h2o混合液的反向液相色谱进行分离纯化,将纯化后的样品冻干,得到约25mg的多肽片段1-mpaa纯品。
[0134]
(7)多肽片段2纯品的制备:取200mg步骤(4)得到的树脂,加入10ml切割试剂(tfa:dodt:h2o=体积比95:2.5:2.5),震荡反应2h,过滤得到黄色透明液体,液体旋蒸仪旋干后,加入约20ml冰乙醚萃取两次,离心后收集沉淀,将样品用20%acn/h2o溶解,通过流动相为含0.1%tfa的acn/h2o混合液的反向液相色谱进行分离纯化,将纯化后的样品冻干,得到约50mg多肽片段2纯品;
[0135]
(8)多肽片段3纯品的制备:取200mg步骤(4)得到的树脂,加入10ml切割试剂(tfa:dodt:h2o=体积比95:2.5:2.5),震荡反应2h,过滤得到黄色透明液体,液体旋蒸仪旋干后,加入约20ml冰乙醚萃取两次,离心后收集沉淀,将样品用20%acn/h2o溶解,通过流动相为含0.1%tfa的acn/h2o混合液的反向液相色谱进行分离纯化,将纯化后的样品冻干,得到约42mg多肽片段3纯品;
[0136]
(9)缓冲盐溶液的配置:配置含6mol/l盐酸胍、0.2mol/l pbs、ph 7.5的缓冲盐溶液(简称溶液a),配置含6mol/l盐酸胍、0.2mol/l pbs、ph 3.0的缓冲盐溶液(简称溶液b),配置含6mol/l盐酸胍、0.2mol/l pbs、ph 6.5的缓冲盐溶液(简称溶液c),缓冲盐溶液a、b、c均使用氮气鼓吹30min,密封保存。
[0137]
(9)负载多肽片段2的spcl树脂的制备:取10mg步骤(2)得到的spcl树脂,加入800μl步骤(9)配置的溶液a,室温震荡溶胀30min,排干,取步骤(7)得到的多肽片段2纯品5.58mg(为spcl树脂负载量的1倍当量),溶解于800μl溶液a,将其于spcl树脂混合,室温下震荡4h,排干溶液,再使用800μl溶液a溶解5.8mg多肽片段2纯品,与spcl树脂混合,室温下震荡4h,排干溶液,使用溶液a洗涤树脂6次,加入15.8mg 2-巯基乙醇(为多肽片段2纯品的20倍当量),反应30min,从而封闭未装载上多肽片段2的spcl树脂的溴基,使用步骤(9)配置的溶液b洗涤6次,得到负载多肽片段2的spcl树脂。为验证多肽片段2在spcl树脂上的成功固定化,取少量上述树脂,加入预先配置的光解溶液(含6mol/l盐酸胍、0.2mol/l pbs、10mm tcep、400mm sem、ph 3.0,氮气鼓吹处理30min),置于4℃下使用365nm的紫外光照射60min,离心,过滤,上清溶液加入1mg tcep反应30min,使用分析hplc和ms对光解反应产物进行检测。
[0138]
(10)负载c端为mpaa的多肽片段2的spcl树脂的制备:称取32.9mg mpaa(为spcl树脂负载量的40倍当量),1.25μl乙酰丙酮(为spcl树脂负载量的2.5倍当量),加入800μl步骤(9)配置的溶液b溶解,与步骤(9)得到的负载多肽片段2的spcl树脂混合,室温下震荡反应6h,使用步骤(9)配置的溶液b洗涤6次,得到负载c端为mpaa的多肽片段2的spcl树脂。为验证多肽片段2在spcl树脂上的成功硫酯化,取少量上述树脂,加入预先配置的光解溶液(含6mol/l盐酸胍、0.2mol/l pbs、10mm tcep、400mm sem、ph 3.0,氮气鼓吹处理30min),置于4
℃下使用365nm的紫外光照射60min,离心,过滤,上清溶液加入1mg tcep反应30min,使用分析hplc和ms对光解反应产物进行检测。
[0139]
(11)负载连接肽1的spcl树脂的制备:称取5.56mg多肽片段3(为spcl树脂负载量的1.3倍当量),32.9mg mpaa(为spcl树脂负载量的40倍当量),14.0mg tcep(为spcl树脂负载量的10倍当量),加入800μl步骤(9)配置的溶液c溶解,与步骤(10)得到的负载c端为mpaa的多肽片段2的spcl树脂混合,室温下震荡反应12h,使用步骤(9)配置的溶液b洗涤6次,得到负载连接肽1的spcl树脂。为验证多肽片段2与片段3在spcl树脂上的成功连接,取少量上述树脂,加入预先配置的光解溶液(含6mol/l盐酸胍、0.2mol/l pbs、10mm tcep、400mm sem、ph 3.0,氮气鼓吹处理30min),置于4℃下使用365nm的紫外光照射60min,离心,过滤,上清溶液加入1mg tcep反应30min,使用分析hplc和ms对光解反应产物进行检测。
[0140]
(12)负载n端为游离半胱氨酸的连接肽1的spcl树脂的制备:取步骤(11)得到的负载连接肽1的spcl树脂,加入800μl 20%哌啶、6mol/l盐酸胍、0.2mol/l pbs、ph 11.0,室温震荡反应30min,脱除n端氨基的fmoc保护基,使用步骤(9)配置的溶液b洗涤6次,加入800μl 0.2mol/l甲氧胺盐酸盐、10mm tcep、6mol/l盐酸胍、0.2mol/l pbs、ph 4.0,室温震荡反应2h,脱除n端半胱氨酸的thz保护基,使用步骤(9)配置的溶液c洗涤6次,得到负载n端为游离半胱氨酸的连接肽1的spcl树脂。
[0141]
(13)负载连接肽2的spcl树脂的制备:称取5.53mg多肽片段1(为spcl树脂负载量的1倍当量),32.9mg mpaa(为spcl树脂负载量的40倍当量),14.0mg tcep(为spcl树脂负载量的10倍当量),加入800μl步骤(9)配置的溶液c溶解,与步骤(12)得到的负载n端为游离半胱氨酸的连接肽1的spcl树脂混合,室温下震荡反应12h,使用步骤(9)配置的溶液b洗涤6次,得到负载连接肽2的spcl树脂。为验证多肽片段1与连接肽1在spcl树脂上的成功连接,取少量上述树脂,加入预先配置的光解溶液(含6mol/l盐酸胍、0.2mol/l pbs、10mm tcep、400mm sem、ph 3.0,氮气鼓吹处理30min),置于4℃下使用365nm的紫外光照射60min,离心,过滤,上清溶液加入1mg tcep反应30min,使用分析hplc和ms对光解反应产物进行检测。
[0142]
(14)负载连接肽2的spcl树脂的紫外光解:取步骤(13)得到的负载连接肽2的spcl树脂的制备,加入500μl预先配置的光解溶液(含6mol/l盐酸胍、0.2mol/l pbs、10mm tcep、400mm sem、ph 3.0,氮气鼓吹处理30min),置于4℃下使用365nm的紫外光照射60min,离心,过滤,上清溶液加入5mg tcep反应30min,对连接肽2粗品溶液进行分离纯化,最终得到1.59mg的连接肽2。
[0143]
步骤(6)获得的多肽片段1-mpaa纯品的反向高效液相色谱与esi-ms的表征,结果如图1所示。
[0144]
步骤(7)获得的多肽片段2纯品的反向高效液相色谱与esi-ms的表征,结果如图2所示。
[0145]
步骤(8)获得的多肽片段3纯品的反向高效液相色谱与esi-ms的表征,结果如图3所示。
[0146]
步骤(9)在制备负载多肽片段2的spcl树脂的过程中,对小量光解反应结束时(60min)的情况进行监测,反向高效液相色谱与重要出峰位置物质的esi-ms的表征,结果如图4所示。
[0147]
步骤(10)在制备负载c端为mpaa的多肽片段2的spcl树脂的过程中,对小量光解反
应初始时(0h),光解反应结束时(60min)的情况进行监测,上述时间点的反向高效液相色谱与重要出峰位置物质的esi-ms的表征,结果如图5所示。
[0148]
步骤(11)在制备负载连接肽1的spcl树脂的过程中,对小量光解反应初始时(0h),光解反应结束时(60min)的情况进行监测,上述时间点的反向高效液相色谱与重要出峰位置物质的esi-ms的表征,结果如图6所示。
[0149]
步骤(13)在制备负载连接肽2的spcl树脂的过程中,对小量光解反应初始时(0h),光解反应结束时(60min)的情况进行监测,上述时间点的反向高效液相色谱与重要出峰位置物质的esi-ms的表征,结果如图7所示。
[0150]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1