一种巴瑞替尼中间体化合物的制作方法
1.本发明属于医药合成技术领域,具体涉及一种巴瑞替尼中间体化合物。
背景技术:
2.巴瑞替尼(baricitinib)是美国礼来制药公司与incyte制药公司合作开发的选择性口服jak1/jak2抑制药,能抑制il-6和il-23等多种炎性细胞因子的细胞内信号传导。2017年2月,巴瑞克替尼获得欧盟批准,作为一种单药或联合甲氨蝶呤,用于对一种或多种疾病修饰抗风湿药物(dmard)缓解不足或不耐受的中度至重度活动性类风湿性关节炎成人患者的治疗。这也是欧盟批准治疗类风湿性关节炎的首个jak抑制剂;2018年6月,美国fda批准了巴瑞克替尼上市,用于治疗罹患中度至重度类风湿关节炎,2022年6月又批准其可作为治疗严重斑秃的口服药。巴瑞替尼化学名为:1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]-3-氮杂环丁烷乙腈,cas号为1187594-09-7,分子式为c
16h17
n7o2s,分子量为371.4,该药物分子中含有吡咯并嘧啶、吡唑、氮杂环丁烷核心结构片段和拥有一个季碳中心,结构式如下:
[0003][0004]
目前,合成巴瑞替尼的方法主要有以下几条路线:
[0005]
路线1:专利wo2009114512及wo2010039939报道由化合物2和带保护基的吡唑硼酸酯(即化合物3)经过suzuki偶联反应得到中间体4,再经盐酸作用脱去吡唑n原子保护基得到关键中间体2;关键中间体1和关键中间体2在1,8-二氮杂双环[5,4,0]十一碳-7-烯(dbu)存在下经过迈克尔加成反应,再脱保护得最终产物巴瑞替尼。当r为特戊酰氧基甲基(pom)时,在lioh溶液中一步脱保护得到巴瑞替尼;当r为2-(三甲基硅基)乙氧基甲基(sem)时,需经libf4和nh4oh两步工序脱除,合成路线如下:
[0006][0007]
该路线中金属钯催化剂的使用,不仅增加了总成本而且存在原料药重金属含量超标的风险;此外,涉及多次脱保护反应,降低了原子经济性。
[0008][0009]
路线2:文献journal of chemical research,2016,40,205-208报道以2-[1-(乙基磺酰基)-3-氮杂环丁亚基]乙腈(即化合物15)为初始原料,在dbu催化下与4-吡唑硼酸频哪醇酯(即化合物13)发生迈克尔加成反应得化合物19;化合物19在重金属钯催化剂存在下与4-氯吡咯并[2,3-d]嘧啶(即化合物9)进行suzuki偶联反应得到巴瑞替尼(1)。
[0010]
该路线尽管只有两步反应,但需采用硅胶柱层析的方式对中间体及终产物进行分离纯化;另外,suzuki偶联反应需在高温下进行48小时,能耗高、工时长。这些因素都严重制约着该反应路线在工业化生产中的实际应用。
[0011][0012]
路线3:专利cn105294699报道以4-吡唑硼酸频哪醇酯(即化合物7)和3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁酯(即化合物8)为起始原料,同样在dbu催化下经迈克尔加成反应制得中间体9,再与化合物10经钯催化suzuki偶联反应制得中间体11;中间体11在酸性条件下同时脱去两分子叔丁氧羰基(boc)得到中间体12;最后,中间体12与乙基磺酰氯经磺酰胺化反应后得到巴瑞替尼(即化合物1)。
[0013]
该路线依然需要通过重金属催化剂实现吡咯并嘧啶环与吡唑环的偶联;化合物11脱boc保护后的产物12含有两个活性氨基位点,在进行磺酰胺化反应时需进行长时间的低温操作,否则会显著增加双磺酰胺化副产物而影响收率与纯度;另外,乙基磺酰氯的使用也增加了原料药中残留基因毒杂质的潜在风险。
[0014][0015]
路线4:专利cn108586465报道以4-氯吡咯并嘧啶(即化合物2)为原料,用二碳酸二叔丁酯(boc2o)对氨基进行保护后得到化合物3,不经纯化,由化合物3与丙烯醛、水合肼在氧气条件下“一锅法”取代、分子内环化制得含吡唑环的化合物4;化合物4与化合物7在乙腈中于四丁基溴化铵(tbab)作用下发生迈克尔加成,再在盐酸溶液中脱氨基保护基得到巴瑞替尼(即化合物1)。
[0016]
该路线在合成化合物4时需要在氧气气氛下回流反应,不仅危险性高且容易产生氧化杂质。我们重复该路线时,发现反应过程中存在化合物3直接和水合肼反应的副产物,
同时化合物3与丙烯醛反应过程中也存在较多1,4-加成副产物(1,2-加成是目标产物),较多结构相似的副产物不仅增加了纯化难度还降低了总收率。
[0017]
因此,针对现有技术中所存在的缺陷,迫切需要一种工艺简便、适合工业化生产、收率高、成本低的巴瑞替尼合成方法,以满足市场需求。
技术实现要素:
[0018]
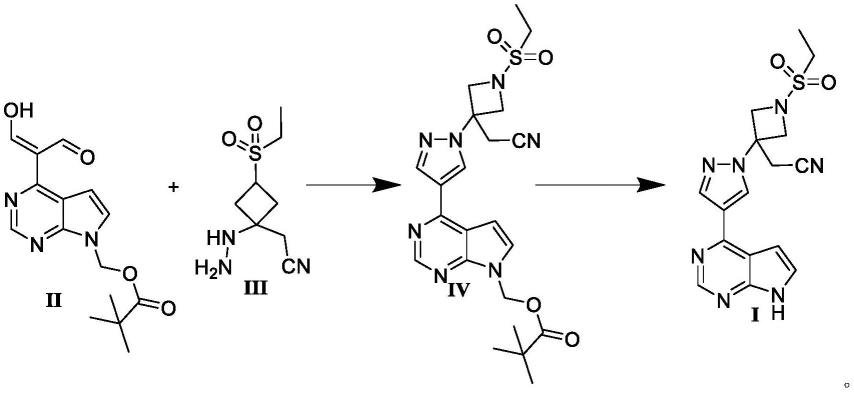
针对以上不足之处,本发明旨在提供一种适合工业化生产巴瑞替尼的合成路线及方法。本路线中成功避免使用重金属催化剂,合成路线简单,反应条件温和、安全性高、操作简便,适合工业放大,具有明显的技术优势。
[0019]
本发明具体通过如下技术方案实现:
[0020]
本发明第一方面提供了一种巴瑞替尼新中间体化合物,其结构如式ii所示:
[0021][0022]
本发明第二方面提供了一种巴瑞替尼中间体化合物ii的制备方法,具体包括如下步骤:将化合物sm-1、化合物sm-2、碱a、碘化钾加入到有机溶剂a中,加热回流至反应结束,反应经后处理得化合物ii:
[0023][0024]
优选地,所述碱a选自碳酸钾、碳酸钠、碳酸氢钠、磷酸氢钠中的一种,其中特别优选碳酸钾。
[0025]
优选地,所述化合物sm-1、化合物sm-2、碱a、碘化钾的投料摩尔比为:1.0:1.0~1.5:1.0~2.0:0.05~0.1,其中特别优选:1.0:1.02:1.2:0.08。
[0026]
优选地,所述有机溶剂a选自四氢呋喃、乙腈、1,2-二氯乙烷,乙醇中的一种或其组合,其中特别优选四氢呋喃。
[0027]
在一优选反应中,反应结束后需要进行后处理,具体操作为:冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii。
[0028]
本发明第三面提供了一种巴瑞替尼新中间体化合物ii用于制备巴瑞替尼的用途,具体包括如下步骤:
[0029]
步骤1:将化合物ii、化合物iii,加入有机溶剂b,加热回流至反应结束,反应经后处理得化合物iv;
[0030]
步骤2:将化合物iv溶于四氢呋喃及甲醇中,搅拌并加入1.0mol/l氢氧化钠水溶液于20-30℃搅拌反应,反应经后处理得巴瑞替尼;
[0031]
合成路线如下:
[0032][0033]
优选地,步骤1所述化合物ii、化合物iii所述投料摩尔比为:1.0:1.0~2.0,其中特别优选1.0:1.1。
[0034]
优选地,步骤1所述有机溶剂b选自乙醇、甲醇、四氢呋喃、乙腈中的一种,其中特别优选乙醇。
[0035]
在一优选反应中,反应结束后需要进行后处理,步骤1具体操作为:反应液冷却,向反应液中加入水,搅拌析晶,抽滤,水洗,真空干燥得化合物iv;步骤2的后处理具体操作为:冷却至0~10℃,用1.0mol/l盐酸水溶液调节ph至中性,继续搅拌1小时后抽滤,乙醇洗涤滤饼,将湿滤饼加入到乙醇/水的混合液中,加热回流后冷却至0~10℃,抽滤,冷乙醇洗涤,真空干燥得化合物i。
[0036]
与现有技术相比,本发明取得的技术效果是:(已修改)
[0037]
1、本发明合成方法的反应步骤少,原子利用率高;
[0038]
2、不使用重金属催化剂,降低原料药重金属残留风险;
[0039]
3、反应条件温和,后处理纯化简单易操作,能耗小工时短,废物排放少,环境友好,适合工业化生产。
具体实施方式
[0040]
下面通过实施例来进一步说明本发明。应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0041]
对本发明得到的化合物结构确证:
[0042]
化合物iii结构表征
[0043][0044]
esi-ms(m/z):219.1[m+h]
+
;1h nmr(400mhz,dmso-d6):δ=4.46(s,4h),3.26(br,3h),3.81(s,2h),3.15(q,2h),1.29(t,3h)ppm。
[0045]
化合物ii结构表征
[0046][0047]
esi-ms(m/z):304.2[m+h]
+
;1h nmr(400mhz,dmso-d6):δ=15.53(s,1h),9.46(s,2h),8.77(s,1h),7.59(d,1h),7.43(d,1h),5.83(s,2h),1.07(s,9h)。
[0048]
化合物iv结构表征
[0049][0050]
esi-ms(m/z):486.5[m+h]
+
;1h nmr(400mhz,cdcl3)δ=8.81(s,1h),8.40(s,1h),8.32(s,1h),7.49(d,1h),6.72(d,1h),6.20(s,2h),4.65(d,2h),4.24(d,2h),3.43(s,2h),3.10(q,2h),1.44(t,3h),1.12(s,9h)ppm。
[0051]
化合物i结构表征
[0052][0053]
esi-ms(m/z)372.5[m+h]
+
;1h nmr(400mhz,dmso-d6)δ=12.15(s,1h),8.94(s,1h),8.72(s,1h),8.49(s,1h),7.63(d,1h),7.09(d,1h),4.62(d,2h),4.25(d,2h),3.71(s,2h),3.24(q,2h),1.26(t,3h)ppm。
[0054]
化合物iii的制备
[0055]
实施例1
[0056]
将2-[1-(乙基磺酰基)-3-氮杂环丁亚基]乙腈(37.24g,200mmol)、80%水合肼(12.50g,200mmol)依次加入至乙腈(100ml)中,搅拌并加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(1.50g,10mmol),于20-30℃搅拌6小时;将反应液倒入到二氯甲烷(200ml)与水(100ml)的混合液中搅拌,静置分液,水相再用二氯甲烷(50ml
×
2)萃取,合并有机层,无水硫酸镁干燥,过滤,收集滤液,减压浓缩得化合物ii,收率98.0%,无需进一步提纯直接用于下一步反应。
[0057]
化合物ii的制备
[0058]
实施例2
[0059]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(16.58g,120mmol)、碘化钾(1.33g,8mmol)加入到thf(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率98.5%,hplc纯度99.72%。
[0060]
实施例3
[0061]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.03g,100mmol)、碳酸钠(12.71g,120mmol)、碘化钾(1.33g,8mmol)加入到thf(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率94.3%,hplc纯度99.66%。
[0062]
实施例4
[0063]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(44.06g,200mmol)、碳酸氢钠(10.08g,120mmol)、碘化钾(1.33g,8mmol)加入到thf(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率95.5%,hplc纯度99.52%。
[0064]
实施例5
[0065]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(13.82g,100mmol)、碘化钾(1.33g,8mmol)加入到乙腈(150ml)中,加热回流6小时;冷却至0
~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率93.8%,hplc纯度99.69%。
[0066]
实施例6
[0067]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(27.64g,200mmol)、碘化钾(1.33g,8mmol)加入到乙醇(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率94.5%,hplc纯度99.52%。
[0068]
实施例7
[0069]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(16.58g,120mmol)、碘化钾(0.83g,5mmol)加入到1,2-二氯乙烷(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率94.8%,hplc纯度99.60%。
[0070]
实施例8
[0071]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(16.58g,120mmol)、碘化钾(1.66g,10mmol)加入到1,2-二氯乙烷(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率95.6%,hplc纯度99.48%
[0072]
实施例9
[0073]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、碳酸钾(16.58g,120mmol)、碘化钾(0.66g,4mmol)加入到thf(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率87.7%,hplc纯度98.92%。
[0074]
实施例10
[0075]
将化合物sm-1(26.77g,100mmol)、化合物sm-2(22.47g,102mmol)、磷酸氢钠(31.23g,220mmol)、碘化钾(1.99g,12mmol)加入到thf(150ml)中,加热回流6小时;冷却至0~10℃,向反应液中加入稀盐酸(1.0mol/l)调节ph至3~4,于20~30℃搅拌2小时后抽滤,冷水洗涤,真空干燥得化合物ii,收率85.3%,hplc纯度98.55%。
[0076]
化合物iv的制备
[0077]
实施例11
[0078]
将化合物ii(15.15g,50mmol)、化合物iii(11.99g,55mmol)于乙醇(150ml)中,加热搅拌过夜,冷却,向反应液中加入水(600ml),搅拌析晶,抽滤,水洗,真空干燥得化合物iv,收率98.0%,hplc纯度99.88%。
[0079]
实施例12
[0080]
将化合物ii(15.15g,50mmol)、化合物iii(10.90g,50mmol)于甲醇(150ml)中,加热搅拌过夜,冷却,向反应液中加入水(600ml),搅拌析晶,抽滤,水洗,真空干燥得化合物iv,收率94.5%,hplc纯度99.72%。
[0081]
实施例13
[0082]
将化合物ii(15.15g,50mmol)、化合物iii(21.80g,100mmol)于四氢呋喃(150ml)中,加热搅拌过夜,冷却,向反应液中加入水(600ml),搅拌析晶,抽滤,水洗,真空干燥得化
合物iv,收率95.1%,hplc纯度99.60%。
[0083]
实施例14
[0084]
将化合物ii(15.15g,50mmol)、化合物iii(26.17g,120mmol)于乙腈(150ml)中,加热搅拌过夜,冷却,向反应液中加入水(600ml),搅拌析晶,抽滤,水洗,真空干燥得化合物iv,收率87.6%,hplc纯度98.55%。
[0085]
巴瑞替尼的制备
[0086]
实施例15
[0087]
将化合物iv(9.71g,20mmol)溶于四氢呋喃(40ml)/甲醇(10ml)混合液中,搅拌并加入1.0mol/l氢氧化钠水溶液(24ml),于20~30℃搅拌2小时;冷却至0~10℃,用1.0mol/l盐酸水溶液调节ph至中性,继续搅拌1小时后抽滤,乙醇洗涤滤饼。将湿滤饼加入到乙醇(40ml)/水(20ml)的混合液中,加热回流半小时后冷却至0~10℃,抽滤,冷乙醇洗涤,真空干燥得化合物1(6.01g,16.18mmol),收率98.9%,hplc纯度99.95%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1