一种以PGF的制作方法
一种以pgf2α
合成中的副产物为原料合成前列地尔的方法
技术领域
1.本发明涉及医药制备领域,特别涉及一种以pgf
2α
合成中的副产物为原料合成前列地尔的方法。
背景技术:
2.前列地尔(alprostadil),简称pge1,是前列腺素系列的重要成员之一。它是一种白色至灰白色结晶粉末。化学名称(1r,2r,3r)-3-羟基-2(e)-(3s)-3-羟基-1-辛烯基-5-氧代环戊烷庚酸,分子式c
20h34
o5。它在临床上主要用于心肌梗死、血栓性脉管炎、闭塞性动脉硬化等症。另外,前列地尔在外脑血管疾病、勃起功能障碍、重症肝炎、急性胰腺炎、慢性胃炎、糖尿病并发症等方面都有疗效和作用。
3.其中,前列地尔的结构式为:
4.前列地尔属于前列腺素类产品,其基本结构特征为:具有一个五元脂环的母体结构及两个侧链,上侧链7个碳、下侧链8个碳。现已报道的人工合成前列地尔的方法主要有两种,第一种方法为corey等人开发的方法,该方法以科里内酯为原料,通过氧化、wittig-horner反应引入下侧链;接着使用(-)-dipcl制备手性醇,并使用tbscl保护;再经过dibal-h低温还原以及wittig反应引入上侧链;最后分别进行氧化、脱保护得到目标产物。具体地,以科里内酯为原料合成前列地尔的合成路径为:
[0005][0006]
第二种方法是以简单的原料出发,经过多步复杂的反应,制备过程中分别使用到对空气和水均敏感的铜、锂等试剂来控制化合物的手性;而且碘甲烷、氢化钠、氰化钾、叔丁
基锂等极易爆或极毒等物料,不易实现工业化生产。其中,第二种方法的合成路径为:
[0007]
技术实现要素:
[0008]
为解决上述背景技术提到的现有技术的不足:本发明提供一种以pgf
2α
合成中的副产物为原料合成前列地尔的方法,其包括以下步骤:
[0009]
s100、对起始原料的15位碳上的羟基进行上硅基保护基反应,并对起始原料的11位碳上的羟基进行选择性保护,生成化合物i;
[0010]
s200、所述化合物i的9位碳上的羟基进行氧化反应,使化合物i的9位碳上的羟基转化为羰基,生成化合物ii;
[0011]
s300、所述化合物ii的5、6位双键进行氢化加成反应,且进行11位碳上的保护基脱除反应,生成到化合物iii;
[0012]
s400、所述化合物iii进行硅基保护基脱除反应,生成前列地尔;
[0013]
其中,所述起始原料包括地诺前列素和/或地诺前列素的5、6位反式异构体,其结构式为化合物i的结构式为化合物ii
的结构式为化合物iii的结构式为
[0014]
在地诺前列素(简称pgf
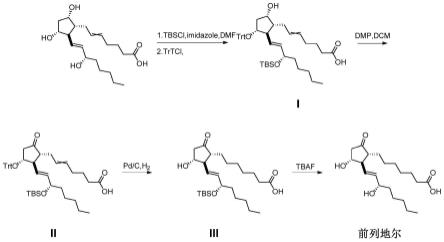
2α
)的合成方法中,其目标产物为地诺前列素,但是合成过程中会产生杂质:地诺前列素的5、6反式异构体杂质化合物b,由于该杂质对生理活性影响较大,因此,药典中均控制其含量必须小于2.5%。但是,地诺前列素的合成路径中,在wittig反应拼拉上侧链反应过程中,由于受温度、空间位阻及官能团等诸多因素的影响,其顺、反异构体的比例接近9:1,再经过多步纯化,分离异构体杂质,pgf
2α
的纯化收率也只有60-70%。因此,其中有接近40%的产品(包含地诺前列素和地诺前列素的5、6反式异构体杂质化合物b),因为异构体含量高而无法使用,只能将该杂质副产品以废弃物的形式处理,造成严重的物料浪费。
[0015]
如果40%的产品能够进行再利用,那么产品的附加值则可以显著的提高,而且也减少废弃物的生成和处理成本。因此本发明从废弃物再利用的角度出发,以绿色合成为指导思想,公开了一种新颖的前列地尔的合成路线,其中以该杂质副产物为起始原料,根据羟基活性和空间位阻等差异,进行选择性保护,接着氧化、选择性氢化加成、脱除保护基等步骤制备高纯度的pge1,由于前列地尔的5,6位是饱和的碳碳单键,通过选择性氢化反应,pgf
2α
以及地诺前列素的5、6反式异构体杂质化合物b的顺、反位的碳碳双键转化为单键并合成目标产物前列地尔。
[0016]
对于该方法的反应机理具体来说,s100中,根据起始原料(地诺前列素和/或地诺前列素的5、6反式异构体杂质化合物b)特性,即根据其15位碳上的羟基具有最高的反应活性,加入硅基保护基进行选择性保护;接着根据空间位阻的不同,再选择性保护11位碳上的羟基,制得化合物i;如图2所示,在pgf
2α
结构式中,9位的羟基与8位碳上的长侧链处于顺式结构,导致其空间位阻很大,而11位碳上的羟基与12位碳上的侧链,处于反式结构空间位阻明显较小,因此s100中可以进行选择性的保护,且上硅基保护基反应和11位碳上的羟基进行选择性保护这两步反应连续投料,不用分离纯化;在s200中,通过氧化反应将化合物i的15位碳上的羟基转化为羰基(即将醇氧化为酮),制得化合物ii;s300中由于化合物ii中15位羟基被保护,导致化合物ii中13、14位双键氢化时位阻变大,氢化加成反应所用催化剂不容易与双键结合,s300中反应可以选择性氢化5、6位碳碳双键,且氢化加成的同时,氢化可以将11位碳上的保护基脱除,制得化合物iii。最后,通过化合物iii脱除硅基保护基反应,即得前列地尔。
[0017]
需要说明的是:地诺前列素(简称pgf
2α
)的结构式为:地诺前列
素的5、6反式异构体杂质化合物b的结构式为:由上述可知,pgf
2α
合成过程中有接近40%的产品,因为异构体含量高而无法使用,这部分包含地诺前列素和地诺前列素的5、6反式异构体杂质化合物b的杂质副产物。本文中将起始原料结构式表达为:表示为起始原料为地诺前列素和/或地诺前列素的5、6反式异构体杂质化合物b。
[0018]
在一些实施例中,所述s100中,所述上硅基保护基反应的步骤为:在溶剂中,在缚酸剂的作用下,羟基保护试剂与起始原料的15位碳上的羟基进行上硅基保护基反应;所述羟基保护试剂为硅醚;所述s100中,通过三苯基氯甲烷与起始原料反应,以选择性保护起始原料的11位碳上的羟基。
[0019]
其中,所述硅醚可优选叔丁基二甲基硅醚、叔丁基二苯基硅醚、三苄基硅醚、三苯基硅醚、三丁基硅醚、三甲基硅醚中的一种或多种组合,以叔丁基二苯基硅醚为较佳选择;傅酸剂可优选三乙胺、二异丙基乙胺、咪唑、4-(n,n-二甲基)-氨基吡啶中的一种或多种组合,以咪唑为较佳选择,所述起始原料与所述傅酸剂的摩尔比优选为1:(2~4),以1:3.2为较佳选择。
[0020]
在一些实施例中,所述起始原料与所述羟基保护试剂的摩尔比为1:(1~1.2),优选1:1;所述起始原料与所述三苯基氯甲烷的摩尔比为1:(1~1.2),优选1:1;所述s1oo中反应温度为0℃~20℃,其中上硅基保护基反应的反应温度优选0℃,对起始原料的11位碳上的羟基进行选择性保护的反应温度优选10℃。
[0021]
在一些实施例中,所述s100中,所述上硅基保护基反应完毕后,于反应体系中直接投入三苯基氯甲烷进行反应。
[0022]
在一些实施例中,在所述s200中,所述化合物i与氧化剂进行9位碳上的羟基氧化反应;所述氧化剂包括三氧化铬的中性溶液、三氧化铬的碱性溶液、戴斯马丁试剂、斯文氧化剂;优选戴斯马丁试剂。
[0023]
在一些实施例中,在所述s200中,所述氧化反应温度为0℃~20℃,优选20℃。
[0024]
在一些实施例中,在所述s300中,所述化合物ii经催化剂催化进行5、6位双键氢化加成反应以及11位碳上的保护基脱除反应;所述催化剂为钯碳、铑碳、铑铝中的一种或多种组合,优选钯碳。
[0025]
在一些实施例中,所述催化剂的重量为所述化合物ii重量的5%~20%,优选钯碳的重量为所述化合物ii重量的10%;所述氢化加成反应的反应温度为-40~-10℃,优选-30℃;且所述氢化反应压力优选0.05mpa。
[0026]
在一些实施例中,在所述s400中,脱保护剂与所述化合物iii混合进行硅基保护基脱除反应;所述脱保护剂包括氢氟酸和/或四丁基氟化铵,优选四丁基氟化铵。
[0027]
在一些实施例中,所述化合物iii与所述脱保护剂的摩尔比为1:(1.5~3),优选1:1.5;所述硅基保护基脱除反应的反应温度为0℃~20℃,优选20℃。
[0028]
在一些实施例中,所述s100中,所述上硅基保护基反应所用溶剂为四氢呋喃、二氧六环、二氯甲烷、n,n-二甲基甲酰胺中的一种或多种组合,优选n,n-二甲基甲酰胺,且其与起始原料的比值为10ml:(0.5~1)g,优选10ml:1g;
[0029]
所述s200中,所述氧化反应所用溶剂为四氢呋喃、二氯甲烷(dcm)、二氧六环中的一种或多种组合,且其与化合物i的比值为10ml:(0.5~1)g,优选二氯甲烷,且其与化合物i的比值为10ml:1g;
[0030]
所述s300中,所述氢化加成反应所用溶剂为醇、四氢呋喃、乙酸乙酯中的一种或多种组合,且其与化合物ii的比值为10ml:(0.5~1)g,优选乙酸乙酯,且其与化合物ii的比值为10ml:1g;
[0031]
所述s400中,所述硅基保护基脱除反应所用溶剂为二氯甲烷、乙腈、乙酸乙酯中的一种或多种组合,优选二氯甲烷,且其与化合物ii的比值优选10ml:1g。
[0032]
与现有的技术相比,本发明提供的一种以pgf
2α
副产物为原料合成前列地尔的方法,具有以下有益效果:
[0033]
本发明提供的该方法,其初始原料为pgf
2α
合成中的副产物,制备成本低,属于回收再利用;该方法的工艺具有原料易得、对设备要求低,步骤简短,操作方便、无需使用到易爆或极毒物料,生产安全性高等特点,容易实现工业化生产;同时,该方法无需重新构建或引入新的手性中心,杂质容易控制,得到的产物光学纯度高。
附图说明
[0034]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
[0035]
图1为本发明提供的实施例1中目标产物前列地尔的合成工艺路线;
[0036]
图2为本发明提供的实施例1中s100反应中羟基选择性保护及双键选择性氢化的机理示意图;
[0037]
图3为本发明提供的实施例1中目标产物前列地尔的1h-nmr图;
[0038]
图4为本发明提供的实施例1中目标产物前列地尔的
13
c-nmr图。
具体实施方式
[0039]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合附图对本发明实施例中的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0040]
本发明提供如下所示实施例和对比例:
[0041]
实施例1:
[0042]
步骤(一)化合物i制备
[0043]
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入pgf
2α
副产物(500g,1.41mol)、咪唑(311g,4.51mol,3.2eq)、dmf(5l),将上述体系冷却至内温≤0℃,并恒温搅拌10min。分批加入叔丁基二苯基氯硅烷(388g,1.41mol,1eq)加至反应体系,控制反应温度不高于0℃。tlc监控反应结束后,再分批加入三苯基氯甲烷(394g,1.41mol,1eq),tlc监控反应结束后,将反应液倒入10l水中,搅拌15分钟,再加入叔丁基甲基醚萃取(2l*3),弃去。水相使用4mol/l的磷酸,酸化至ph=5,再使用叔丁基甲基醚萃取(2l*3),有机相合并后分别用饱和氯化钠(1l)溶液洗涤、无水硫酸钠干燥、浓缩,得淡黄色化合物i(821g,收率82%),可直接应用于下步反应。需要说明的是,使用的保护原料是氯硅烷,反应后生成硅醚,硅醚作为羟基保护剂。
[0044]
步骤(二)化合物ii制备
[0045]
在装有温度计、机械搅拌及滴液漏斗的10l的反应釜中,顺次加入化合物i(821g,1.15mol,1eq)和二氯甲烷(8l)。开启搅拌,将上述体系冷却至内温≤10℃,接着将下戴斯马丁试剂dmp(539g,1.27mol,1.1eq)分批加入至反应体系,加完后自然回温,tcl检测反应完成后,倒入水中。室温搅拌1小时后静置分层。水相再使用二氯甲烷萃取(2l)。合并有机相,分别经过饱和氯化钠(5l)洗涤、无水硫酸钠干燥、浓缩后得淡黄色,化合物ii(763g,收率93%)。
[0046]
步骤(三)化合物iii制备
[0047]
在10l的氢化反应釜中,分别加入化合物ii(763g,1.07mol,1eq)、10%钯碳(76g)及乙酸乙酯(7.6l)。将反应釜内温度降至-35℃后,开始氮气制备三次,再使用氢气置换三次后,保持内压0.05mpa。hplc检测反应结束后,氮气置换三次,过滤,并使用乙酸乙酯洗涤滤饼。滤液无水硫酸钠干燥、浓缩后得粗品800g,再加入叔丁基甲基醚2l,打浆、过滤、洗涤、干燥,得化合物iii(426g,收率85%)。
[0048]
步骤(四)化合物iv制备
[0049]
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入化合物iii(426g,0.91mol,1eq),二氯甲烷(5l)。然后将上述混合体系,冷却至内温≤10℃,并恒温搅拌10min。再将四丁基氟化铵(356g,1.36mol,1.5eq)缓慢加至反应体系,控制反应温度不高于10℃。反应结束后,体系中加入饱和氯化钠(5000ml)淬灭,分层后水相使用二氯甲烷萃取(1l*2),有机相弃去。水相使用4mol/l的磷酸,酸化至ph=5,再使用叔丁基甲基醚萃取(2l*3),有机相合并后分别用饱和氯化钠(1l)溶液洗涤、无水硫酸钠干燥、浓缩,得淡黄色固体粗品。将上述粗品加入至1l的乙酸乙酯和20g的活性碳中,40℃加热10分钟,趁热过滤,滤饼再使用乙酸乙酯洗涤。滤液缓慢冷却至0℃,析晶、过滤、洗涤、干燥,得前列地尔241g,收率75%。熔点115-116℃,[α]d20=-52。(c=0.25,c2h5oh);制得的前列地尔成品的核磁共振表征结果如图3-4所示,具体数据如下:
[0050]
hnmr(500mhz,cd3od)δ=5.44-5.51(m,2h),5.04(bs,1h),4.60(bs,1h),3.87-3.94(bs,2h),2.55(dd,1h,j=6.0,15.0hz,1h),2.18-2.22(m,1h),2.15-2.17(m,2h),1.97-2.06(m,2h),1.20-1.47(m,18h),0.85(t,j=5.5hz,3h);13cnmr(125mhz,cd3od)δ:216.19,174.92,136.70,130.69,71.54,71.39,53.90,53.77,47.02,37.90,34.11,31.78,29.42,28.84,27.69,26.63,25.15,24.91,22.62,14.35。hrms-esi,m/z for c20h34o5[m+h]+calc 353.2327,found 353.2324.
[0051]
实施例2
[0052]
步骤(一)化合物i制备
[0053]
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入pgf
2α
副产物(500g,1.41mol)、咪唑(311g,4.51mol,3.2eq)、dmf(5l),将上述体系冷却至内温≤0℃,并恒温搅拌10min。分批加入叔丁基二甲基氯硅烷(211g,1.41mol,1eq)加至反应体系,控制反应温度不高于0℃。tlc监控反应结束后,再分批加入三苯基氯甲烷(394g,1.41mol,1eq),tlc监控反应结束后,将反应液倒入10l水中,搅拌15分钟,再加入叔丁基甲基醚萃取(2l*3),弃去。水相使用4mol/l的磷酸,酸化至ph=5,再使用叔丁基甲基醚萃取(2l*3),有机相合并后分别用饱和氯化钠(1l)溶液洗涤、无水硫酸钠干燥、浓缩,得淡黄色化合物i(751g,收率75%),可直接应用于下步反应。
[0054]
步骤(二)化合物ii制备
[0055]
在装有温度计、机械搅拌及滴液漏斗的20l的反应釜中,顺次加入二氯甲烷(8l)、pcc(339g,1.58mol,1.5eq)和醋酸钠(264g,3.15mol,3eq)开启搅拌,将上述体系冷却至内温≤10℃,接着化合物i(751g,1.05mol,1eq)和二氯甲烷(1l)。滴加至反应体系,加完后自然回温,tcl检测反应完成后,倒入水中。室温搅拌1小时后静置分层。水相再使用二氯甲烷萃取(2l)。合并有机相,分别经过饱和氯化钠(5l)洗涤、无水硫酸钠干燥、浓缩后得淡黄色,化合物ii(690g,收率92%)。
[0056]
步骤(三)化合物iii制备
[0057]
在10l的氢化反应釜中,分别加入化合物ii(690g,0.97mol,1eq)、10%钯碳(70g)及乙酸乙酯(7l)。将反应釜内温度降至-35℃后,开始氮气制备三次,再使用氢气置换三次后,保持内压0.05mpa。hplc检测反应结束后,氮气置换三次,过滤,并使用乙酸乙酯洗涤滤饼。滤液无水硫酸钠干燥、浓缩后得粗品800g,再加入叔丁基甲基醚2l,打浆、过滤、洗涤、干燥,得化合物iii(391g,收率86%)。
[0058]
步骤(四)化合物iv制备
[0059]
在装有恒压滴液漏斗、温度计及机械搅拌的10l三口瓶中分别加入化合物iii(391g,0.83mol,1eq),二氯甲烷(5l)。然后将上述混合体系,冷却至内温≤10℃,并恒温搅拌10min。再将四丁基氟化铵(325g,1.24mol,1.5eq)缓慢加至反应体系,控制反应温度不高于10℃。反应结束后,体系中加入饱和氯化钠(5000ml)淬灭,分层后水相使用二氯甲烷萃取(1l*2),有机相弃去。水相使用4mol/l的磷酸,酸化至ph=5,再使用叔丁基甲基醚萃取(2l*3),有机相合并后分别用饱和氯化钠(1l)溶液洗涤、无水硫酸钠干燥、浓缩,得淡黄色固体粗品。将上述粗品加入至1l的乙酸乙酯和20g的活性碳中,40℃加热10分钟,趁热过滤,滤饼再使用乙酸乙酯洗涤。滤液缓慢冷却至0℃,析晶、过滤、洗涤、干燥,得前列地尔217g,收率74%。制得的前列地尔成品进行核磁共振表征,1h-nmr图和
13
c-nmr图数据表明其为前列地尔,在此只列出实施例1中的相关图谱,其他实施例2的谱图和测试过程此处不再累述。通过上述实施例的测试结果可知:
[0060]
本发明实施例所示合成方法原料为pgf
2α
合成中的副产物,其初始原料廉价易得,且其工艺简单、路线简短、对设备的要求低,且本发明实施例所示合成方法中无危险性试剂和操作,容易实现大规模工业化生产;同时,其无需重新构建或引入新的手性中心,中间体纯化方法简单、杂质容易控制,得到的产物pge1光学纯度高,实施例中前列地尔的反应收率
可达到50%以上。
[0061]
相对传统的前列地尔合成方法,本发明包括至少以下发明构思和创新点:本发明的起始原料与传统合成方法不同,本发明是以废弃物为初始原料开始合成,针对该初始原料,第一步骤利用空间位阻和官能团反应活性选择保护羟基;并且在氢化加成反应步骤,5、6位双键进行氢化加成反应与11位碳上的保护基脱除反应同时进行,反应收率高;在化合物iii进行硅基保护基脱除反应中,弱碱性脱15位硅醚保护,无11位消除副反应;本发明合成方法具有反应收率高,合成化合物产物的15位异构体少,光学纯度高等优势。
[0062]
综上所述,本发明公开了一种以pgf
2α
副产物为原料,低成本合成前列地尔的新方法。该方法从废弃物再利用的角度出发,以绿色合成为指导思想,公开了一种新颖的合成路线。其具有原料易得、步骤短、操作简单和对设备要求低等特点;同时中间体纯化方法简单、杂质容易控制,得到的产物光学纯度高等特性。
[0063]
需要说明的是:
[0064]
本文中采用“~”表示数值范围,该表达方式的表示范围内包含两个端点值。
[0065]
本文中,戴斯马丁氧化反应中使用的戴斯马丁试剂包括dmp,所述“dmp”为本领域常规的缩写简称,又名戴斯-马丁高碘烷,dess-martin periodinane;英文名称1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1h)-one,并非指代邻苯二甲酸二甲酯。
[0066]
对于斯文氧化试剂(swan氧化试剂),本领域中用二甲亚砜(dmso)-乙二酰氯(草酰氯)或三氟乙酸酐体系对醇的氧化称为斯文氧化,斯文氧化试剂即指的二甲亚砜(dmso)-乙二酰氯(草酰氯)体系。
[0067]
图1中,反应物料名称采用本领域常用缩写,tbscl为叔丁基二甲基氯硅烷、trtcl为三苯基氯甲烷,dcm为二氯甲烷、pd/c为钯碳,tbaf为四丁基氟化铵。
[0068]
本文中将起始原料结构式表达为:表示为起始原料为地诺前列素和/或地诺前列素的5、6反式异构体杂质化合物b。与之同理,根据初始原料构型,化合物i为顺、反异构体中的一种或两种组合,其结构式为化合物ii为顺、反异构体中的一种或两种组合,其结构式为
[0069]
上述实施例中的具体参数或一些常用试剂或原料,为本技术构思下的具体实施例或优选实施例,而非对其限制;本领域技术人员在本技术构思及保护范围内,可以进行适应性调整。
[0070]
此外,若无特殊说明,所采用的原料也可以为本领域常规市售产品、或者由本领域常规方法制备得到;即本实施例中所用试剂、仪器未注明生产厂家等信息,均为可以通过市场购买获得的常规产品。
[0071]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1