一种手性离子液体及其应用
1.本发明属于化工产品技术领域,具体涉及一种手性离子液体及其应用。
背景技术:
2.手性是自然界中生物体的基本属性,在手性环境中,互为镜像的对映异构体往往展现出某些不同的特性,例如反应速度或生理作用等。特对是对于制药行业来说,具有手性中心的活性药物成分因构型不同有时会导致不同的临床反应、药理活性和毒理性。苏氨酸(threonine)又称β-羟基-α-氨基丁酸,具有两种单一对映体l-型和d-型,其中l-苏氨酸(l-thr)是一种必需的氨基酸,主要用于医药、饲料添加剂等方面;对于动物来说,l-苏氨酸可改善氨基酸消化率低的饲料原料的营养价值,提高低能量饲料生产性能;对于人类来说,其用于消化溃疡的辅助治疗,也可治贫血及心绞痛、主动脉炎和心功能不全等心血管系统疾患,而d-苏氨酸则没有生物学活性,d-苏氨酸或其外消旋体的存在将极大地限制相关药物的疗效。因此开发一种产品纯度高,操作简单,绿色高效的外消旋苏氨酸的拆分技术是十分重要的。
3.申请号为cn200710185671.x的中国专利公开了一种dl-n-乙酰苏氨酸酯直接生物催化水解拆分制备l-和d-苏氨酸的新方法。但此方法首先对菌种的筛选、处理工艺、保存等条件较为严苛;其次利用酶拆分苏氨酸的过程中需要控制温度、溶液ph值、加入金属离子以及助溶剂等条件过多;最后获得的含有产品水溶液仍需要使用离子交换树脂处理才能获得产品,后处理要求较高。总之此方法工艺繁琐,过程耗时,成本较高。
4.申请号为cn201710542082.6的中国专利公开了一种在饱和外消旋苏氨酸溶液中,同时加入一种与染料分子复合的聚合物以及晶种,通过降温后析出不同颜色的苏氨酸对映体晶体进行分级结晶拆分的方法。但此方法首先选用的聚合物合成步骤较为复杂,技术要求、成本高;其次在使用聚合物作为抑制剂的同时仍然需要加入晶种;最后产品纯度ee值小于99%,纯度不高。总之此方法技术要求较高,纯度较低,不利于工业化。
5.申请号为cn201410152313.9的中国专利公开了一种新型托品醇类氨基酸阴离子型手性离子液体的制备、固定化及其拆分dl-苯丙氨酸和dl-色氨酸的方法。该专利中,由托品醇与ch3(ch2)nbr(n=1-7)加热反应获得托品醇溴化物,将其通过强碱性阴离子树脂,所得溶液经浓缩后,加入l-脯氨酸,最终获得黄色粘稠液体产品,再将此离子液体与盐溶液形成双水相液液萃取拆分dl-色氨酸和苯丙氨酸。该专利中将手性离子液体应用于液液萃取拆分,但拆分产品纯度不高。
6.综上,上述专利中的外消旋苏氨酸的酶拆分以及加入晶种和聚合物的分级结晶拆分方法存在工艺繁琐和纯度低的缺陷。因此,一种工艺简单和纯度高的外消旋体的拆分方法是有必要的。
技术实现要素:
7.针对以上问题,本发明目的在于提供一种手性离子液体,可以基于该手性离子液
体对外消旋苏氨酸进行拆解得到l-苏氨酸,该方法拆解得到的l-苏氨酸ee值高,纯度高,工艺简洁。
8.为了达到上述目的,本发明可以采用以下技术方案:
9.本发明一方面提供一种手性离子液体,其制备方法包括:(1)将1-己基-3-甲基咪唑溴盐与717阴离子树脂进行离子交换得[c6mim]oh水溶液;(2)将[c6mim]oh水溶液与过量手性酸进行中和反应得混合物,其中,手性酸选自l-2-氨基丁酸、d-2-氨基丁酸、l-苹果酸或d-苹果酸;(3)将混合物除水后,加入有机溶剂使得手性酸析出,过滤除去固体得到滤液1;(4)将滤液1蒸发,干燥得手性离子液体。
[0010]
本发明另一方面提供一种拆分外消旋苏氨酸的方法,其包括:(1)将上述的手性离子液体与水混合得潜溶剂;(2)将过量的外消旋苏氨酸在潜溶剂中混合至固液平衡后得饱和溶液;(3)将饱和溶液降温结晶,过滤得l-苏氨酸。
[0011]
本发明有益效果包括:
[0012]
(1)本发明提供的手性离子液体,其合成步骤简单、手性识别效果优异且可回收循环使用;而且通过该手性离子液体与水形成的潜溶剂,解决了离子液体高粘度带来传质影响,同时提高了外消旋苏氨酸的溶解度;
[0013]
(2)本发明提供的拆分外消旋苏氨酸的方法利用本发明中的手性离子液体进行作为手性溶剂结晶拆分外消旋苏氨酸的过程中,异手性的离子液体与苏氨酸对映体之间有着更强作用力,导致手性离子液体抑制与其异手性的对映体成核,使得与其同手性的对映体优先成核析出。操作过程中仅通过手性离子液体即可完成拆分,无需额外加入晶种,进行1-2次循环结晶拆分过程即可获得高纯度苏氨酸对映体ee值大于99%,操作简单,分离效果显著,工艺循环,成本低廉,避免了已有技术中(酶拆分以及加入晶种和聚合物的分级结晶拆分)工艺复杂、纯度较低等问题。
附图说明
[0014]
图1为实施例1手性离子液体[hmim][l-2-aba]的核磁图;
[0015]
图2为实施例1手性离子液体[hmim][l-2-aba]的热重分析图;
[0016]
图3为实施例2手性离子液体[hmim]2[l-ma]的核磁图;
[0017]
图4为实施例2手性离子液体[hmim]2[l-ma]的热重分析图;
[0018]
图5为实施例1制备的[hmim][l-2-aba]不同浓度潜溶剂中外消旋苏氨酸的溶解度图;
[0019]
图6为实施例2制备的[hmim]2[l-ma]不同浓度潜溶剂中外消旋苏氨酸的溶解度图;
[0020]
图7为标准品消旋苏氨酸的hplc色谱图;
[0021]
图8为实施例5产品的hplc色谱图;
[0022]
图9为实施例6产品的hplc色谱图;
[0023]
图10为实施例7产品的hplc色谱图;
[0024]
图11为实施例8产品的hplc色谱图;
[0025]
图12为实施例9产品的hplc色谱图;
[0026]
图13为实施例10产品的hplc色谱图。
具体实施方式
[0027]
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0028]
本文中使用的术语仅用于描述特定实施例,并且无意于限制本公开。除非在上下文中具有明显不同的含义,否则单数形式的表达包括复数形式的表达。如本文所使用的,应当理解,诸如“包括”、“具有”、“包含”之类的术语旨在指示特征、数字、操作、组件、零件、元件、材料或组合的存在。在说明书中公开了本发明的术语,并且不旨在排除可能存在或可以添加一个或多个其他特征、数字、操作、组件、部件、元件、材料或其组合的可能性。如在此使用的,根据情况,“/”可以被解释为“和”或“或”。
[0029]
本发明一实施例提供一种手性离子液体,其制备方法包括:(1)将1-己基-3-甲基咪唑溴盐与717阴离子树脂进行离子交换得[c6mim]oh水溶液;(2)将[c6mim]oh水溶液与过量手性酸进行中和反应得混合物,其中,手性酸选自l-2-氨基丁酸、d-2-氨基丁酸、l-苹果酸或d-苹果酸;(3)将混合物除水后,加入有机溶剂使得手性酸析出,过滤除去固体得到滤液1;(4)将滤液1蒸发,干燥得手性离子液体。
[0030]
需要说明的是,步骤(4)中,制备得到的手性离子液体为1-己基-3-甲基咪唑l-2-氨基丁酸盐([hmim][l-2-aba])、1-己基-3-甲基咪唑d-2-氨基丁酸盐([hmim][d-2-aba])、1-己基-3-甲基咪唑l-苹果酸盐([hmim]2[l-ma])和1-己基-3-甲基咪唑d-苹果酸盐([hmim]2[d-ma])。
[0031]
另外,离子液体(ionicliquids,ils)是指由有机阳离子和无机或有机阴离子构成的、在室温或室温附近温度下呈液态的盐类。手性离子液体(chiralionicliquids,cils)是功能化离子液体的一种,具有手性材料和液体材料的双重特征。相较于传统手性溶剂,手性离子液体有过冷的趋势,这使其具有相当宽的液相操作温度范围,避免了手性溶剂熔点过高带来的问题。
[0032]
另外,上述手性离子液体的制备中,阳离子采用咪唑类溴盐1-己基-3-甲基咪唑溴盐提供,阴离子由一元酸l-2-氨基丁酸,d-2-氨基丁酸、二元酸l-苹果酸和d-苹果酸提供,通过1-己基-3-甲基咪唑溴盐的经过强碱性阴离子树脂后的氢氧化物和手性酸进行酸碱中和反应获得手性离子液体。
[0033]
在一些具体实施例中,上述手性离子液体的制备方法中,步骤(2)中,中和反应在冰浴中进行。
[0034]
在一些具体实施例中,上述手性离子液体的制备方法中,步骤(3)中,将混合物除水的方法包括:将混合物在323.15k-333.15k下旋转蒸发1.8h-2.2h,比如在333.15k下旋转蒸发2h。
[0035]
在一些具体实施例中,上述手性离子液体的制备方法中,步骤(3)中,将混合物除水后,在273.15k-276.15k下在混合物中加入有机溶剂使得手性酸析出;其中,有机溶剂选自乙腈和甲醇的混合溶剂或乙腈和乙醚的混合溶剂。需要说明的是,在混合过程中,可以加入有机溶剂溶解的同时进行剧烈搅拌。
[0036]
在一些具体实施例中,上述手性离子液体的制备方法中,步骤(3)中,乙腈和甲醇的混合溶剂中,乙腈和甲醇的体积比8-10:1,优选9:1;乙腈和乙醚的混合溶剂中,乙腈和乙
醚的体积比8-10:1,优选9:1。需要说明的是,乙腈和甲醇的混合溶剂对应除去手性酸l-和d-2-氨基丁酸;乙腈和乙醚的混合溶剂对应除去手性酸l-和d-苹果酸。
[0037]
在一些具体实施例中,上述手性离子液体的制备方法中,步骤(4)中,蒸发可以选择旋转蒸发,干燥选择真空干燥。
[0038]
本发明另一实施例提供一种拆分外消旋苏氨酸的方法,其包括:(1)将上述的手性离子液体与水混合得潜溶剂;(2)将过量的外消旋苏氨酸在潜溶剂中混合至固液平衡后得饱和溶液;(3)将饱和溶液降温结晶,过滤得l-苏氨酸。
[0039]
需要说明的是,上述拆分外消旋苏氨酸过程中,手性溶剂会给整个溶液系统提供手性环境,手性溶剂与手性溶质之间产生选择性的相互作用,这有助于区分两种单一对映体,从而将外消旋体成功拆分。
[0040]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法中,步骤(1)中,潜溶剂中手性离子液体的浓度为10wt%-70wt%。
[0041]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法中,步骤(2)中,混合至达到固液平衡的时间为12h-36h。
[0042]
需要说明的是,达到固液平衡时间由手性离子液体水溶液的浓度决定,当其小于30wt%时,搅拌和静置时间分别为12小时;当其大于等于30wt%小于60wt%时,搅拌和静置时间分别为24小时;当其大于等于60wt%时,搅拌和静置时间分别为36小时。并且搅拌和静置时的温度均为313.15k。
[0043]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法中,步骤(3)中,降温结晶时,搅拌转速为340rpm-360rpm,降温速率为0.05k/min-0.15k/min;比如,搅拌转速可以为350rpm,降温速率可以为0.1k/min。
[0044]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法中,步骤(3)中,在饱和溶液降温结晶时检测ee值,ee值最高时进行过滤。
[0045]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法中,步骤(3)中,过滤得母液;将母液蒸发除水,然后用有机溶剂溶解,过滤除去苏氨酸得滤液2;将滤液2蒸发除去有机溶剂,干燥得手性离子液体。在一些具体实施例中,将母液中手性离子液体回收的方式为:母液旋转蒸发除水后,在冰浴的条件下,将所得的混合物用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去苏氨酸。收集滤液并旋转蒸发除去有机溶剂,放入333.15k的真空烘箱干燥48小时获得手性离子液体可再次使用。
[0046]
需要说明的是,上述拆分外消旋苏氨酸的方法中,在30wt%[hmim]2[l-ma]和[hmim][l-2-aba]与水的潜溶剂中,结晶拆分循环得到高纯度产品的次数分别为1次和2次。
[0047]
在一些具体实施例中,上述拆分外消旋苏氨酸的方法可以包括以下步骤:
[0048]
(1)配置不同浓度的上述手性离子液体与水的潜溶剂,作为结晶拆分的手性溶剂体系;过量的外消旋苏氨酸溶解在手性离子液体水溶液中,并在313.15k下搅拌12~36小时后,静置12h-36h,使其达到固液平衡。
[0049]
(2)取出313.15k下的饱和溶液,放入连有程序控温装置的313.15k的结晶器中,以350rpm的搅拌转速,0.2k/min的降温速率开始降温,在处析出产品后开始取样。每相隔5min进行一次取样,共取样10次,过滤获得固体产品;用高效液相色谱检测15次样品的光学纯度,确定产品ee值最高的时间点;
[0050]
(3)重复步骤(1)和(2),在进行步骤(2)时只在产品ee值最高的时间点取样过滤获得产品。
[0051]
(4)将母液旋转蒸发除水后,用有机溶剂溶解所得的混合物,过滤除去苏氨酸。收集滤液、旋转蒸发、真空干燥获得手性离子液体,其可再次循环使用;
[0052]
(5)将步骤(3)中的固体产品重复步骤(1)、(2)和(3);循环1-2次操作即可获得高纯度产品。
[0053]
为了更好地理解本发明,下面结合具体示例进一步阐明本发明的内容,但本发明的内容不仅仅局限于下面的示例。
[0054]
实施例1制备手性离子液体[hmim][l-2-aba]和[hmim][d-2-aba]
[0055]
称取2.47g(0.01mol)1-己基-3-甲基咪唑溴盐([c6mim]br),并用10ml超纯水溶解;将其通过预处理过的717强碱性阴离子树脂进行离子交换获得[c6mim]oh水溶液;其次在冰浴的条件下,将[c6mim]oh水溶液通过恒压滴液漏斗逐滴缓慢加入装有1.03g(0.01mol)l-2-氨基丁酸或d-2-氨基丁酸和20ml超纯水的烧瓶中,并搅拌反应12h,反应结束后将溶液旋转蒸发除去水;接着在冰浴的条件下,将获得的混合物用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去过量的l-2-氨基丁酸或d-2-氨基丁酸,将滤液再次旋转蒸发除去溶剂;最后将获得的液体放入353.15k的真空烘箱中干燥48h获得手性离子液体。
[0056]
将制备得到的[hmim][l-2-aba]手性离子液体进行核磁分析,结果如图1所示,1hnmr(400mhz,dmso-d6)δ9.44(d,j=1.7hz,1h),7.81(t,j=1.8hz,1h),7.74(t,j=1.8hz,1h),4.17(t,j=7.2hz,2h),3.87(s,3h),1.78(p,j=7.4hz,2h),1.54(dtd,j=14.9,7.4,4.8hz,1h),1.36
–
1.15(m,8h),0.90
–
0.83(m,3h),0.79(t,j=7.4hz,3h),证明了手性离子液体[hmim][l-2-aba]成功合成;
[0057]
将制备得到的[hmim][l-2-aba]手性离子液体进行热重分析,结果如图2所示,手性离子液体[hmim][l-2-aba]的分解温度为476.26k,作为溶剂其具有良好的热稳定性;
[0058]
另外,l-型离子液体与其对应的d-型离子液体仅存在旋光性差别,在物理特征上两者保持一致,故手性离子液体[hmim][l-2-aba]和[hmim][d-2-aba]均成功合成,均具有良好的热稳定性。
[0059]
实施例2制备手性离子液体1-己基-3-甲基咪唑l-苹果酸盐([hmim]2[l-ma])和1-己基-3-甲基咪唑d-苹果酸盐([hmim]2[d-ma])
[0060]
称取2.47g(0.01mol)1-己基-3-甲基咪唑溴盐([c6mim]br),并用10ml超纯水溶解;将其通过预处理过的717强碱性阴离子树脂进行离子交换获得[c6mim]oh水溶液;其次在冰浴的条件下,将[c6mim]oh水溶液通过恒压滴液漏斗逐滴缓慢加入装有0.67g(0.005mol)l-苹果酸或d-苹果酸和20ml超纯水的烧瓶中,并搅拌反应12h,反应结束后将溶液旋转蒸发除去水;接着在冰浴的条件下,将获得的混合物用体积比9:1的乙腈和乙醚溶解并剧烈搅拌,过滤除去过量的l-苹果酸或d-苹果酸,将滤液再次旋转蒸发除去溶剂。最后将获得的液体放入353.15k的真空烘箱中干燥48h获得手性离子液体;
[0061]
将制备得到的手性离子液体[hmim]2[l-ma]进行核磁分析,结果如图3所示,1hnmr(400mhz,dmso-d6)δ9.27(d,j=1.7hz,2h),7.81(t,j=1.8hz,2h),7.74(t,j=1.8hz,2h),4.17(t,j=7.2hz,4h),4.02(dd,j=8.3,5.3hz,1h),3.86(s,6h),2.58
–
2.52(m,1h),2.43
–
2.33(m,1h),1.78(p,j=7.4hz,4h),1.33
–
1.18(m,12h),0.91
–
0.81(m,6h),证明了手性离
子液体[hmim]2[l-ma]成功合成;
[0062]
将制备得到的手性离子液体[hmim]2[l-ma]进行热重分析,结果如图4所示,手性离子液体[hmim]2[l-ma]的分解温度为495.17k,作为溶剂其具有良好的热稳定性。
[0063]
另外,本发明中的l-型离子液体与其对应的d-型离子液体仅存在旋光性差别,在物理特征上两者保持一致,故手性离子液体[hmim]2[l-ma]和[hmim]2[d-ma]均成功合成,具有良好的热稳定性。
[0064]
实施例3不同浓度潜溶剂溶解度
[0065]
将实施例1制备的[hmim][l-2-aba]与水配置成不同浓度(10wt%、30wt%、50wt%和70wt%)的潜溶剂,不同浓度潜溶剂中的溶解度图如图5所示;将实施例2制备的[hmim]2[l-ma]与水配置成不同浓度(10wt%、30wt%、50wt%和70wt%)的潜溶剂,不同浓度潜溶剂中的溶解度图如图5所示;由图5和图6中可以看出,相较于外消旋苏氨酸在纯水中的溶解度,手性离子液体与水形成的潜溶剂在30wt%的比例时溶解度最大且超过了在纯水中的溶解度,同时可以看出外消旋苏氨酸在30wt%[hmim]2[l-ma]中的溶解度略大于在30wt%[hmim][l-2-aba]。
[0066]
实施例4外消旋苏氨酸hplc色谱图
[0067]
本发明实施例中,对标准品外消旋苏氨酸进行高效液相色谱(hplc)检测,hplc检测方法为:大赛璐crownpakcr(+)手性柱,流动相为ph=1.5的高氯酸溶液,洗脱速率为0.2ml/min,洗脱时间为20min,柱温箱温度为278.15k,检测器波长200nm。检测结果如图7所示,图中l-苏氨酸的保留时间为10.70min,d-苏氨酸的保留时间为8.15min。
[0068]
实施例5利用实施例1制得的手性离子液体拆分外消旋苏氨酸
[0069]
(1)配置10wt%[hmim][l-2-aba]与水的潜溶剂,以实现低粘度的手性溶剂体系。过量的外消旋苏氨酸溶解在10wt%[hmim][l-2-aba]的水溶液中,并在313.15k下搅拌12小时后,静置12小时,使其达到固液平衡;
[0070]
(2)取出313.15k下的饱和溶液,放入连有程序控温装置的313.15k的结晶器中,以350rpm的搅拌转速,0.1k/min的降温速率开始降温,析出产品后开始取样,每相隔5min进行一次取样,共取样15次,过滤获得固体产品。用高效液相色谱检测(方法同实施例4)15次样品的光学纯度,确定产品ee值最高的时间点;
[0071]
(3)重复步骤(1)和(2),在进行步骤(2)时只在产品ee值最高的时间点取样过滤获得产品;ee值最高时的hplc色谱图如图8所示,在保留时间9.86min处存在l-thr,l-thr的峰面积百分比为66.45;在保留时间7.21min处存在d-thr,d-thr的峰面积百分比为33.54,由此可得,l-thr对映体过量,ee值为32.92%;
[0072]
(4)将母液旋转蒸发除水,在冰浴的条件下,将所得的混合物用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去苏氨酸。收集滤液并旋转蒸发除去有机溶剂,放入333.15k的真空烘箱干燥48小时获得手性离子液体可再次使用。
[0073]
实施例6利用实施例1制得手性离子液体拆分外消旋苏氨酸
[0074]
(1)配置30wt%[hmim][l-2-aba]与水的潜溶剂,以实现低粘度的手性溶剂体系,过量的外消旋苏氨酸溶解在30wt%[hmim][l-2-aba]的水溶液中,并在313.15k下搅拌24小时后,静置24小时,使其达到固液平衡;
[0075]
(2)取出313.15k下的饱和溶液,放入连有程序控温装置的313.15k的结晶器中,以
350rpm的搅拌转速,0.1k/min的降温速率开始降温,析出产品后开始取样,每相隔5min进行一次取样,共取样15次,过滤获得固体产品。用高效液相色谱检测(方法同实施例4)15次样品的光学纯度,确定产品ee值最高的时间点;
[0076]
(3)重复步骤(1)和(2),在进行步骤(2)时只在产品ee值最高的时间点取样过滤获得产品;ee值最高时的hplc色谱图如图9所示,在保留时间9.47min处存在l-thr,l-thr的峰面积百分比为79.87;在保留时间7.13min处存在d-thr,d-thr的峰面积百分比为20.13,由此可得,l-thr对映体过量,ee值为59.74%;
[0077]
(4)将母液旋转蒸发除水,在冰浴的条件下,将所得的混合物用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去苏氨酸。收集滤液并旋转蒸发除去有机溶剂,放入333.15k的真空烘箱干燥48小时获得手性离子液体可再次使用。
[0078]
实施例7利用实施例1制备得到的手性离子液体拆分外消旋苏氨酸
[0079]
(1)将实施例6中步骤(3)中的固体产品过量溶于30wt%[hmim][l-2-aba]与水的潜溶剂,并在313.15k下搅拌24小时后,静置24小时,使其达到固液平衡;
[0080]
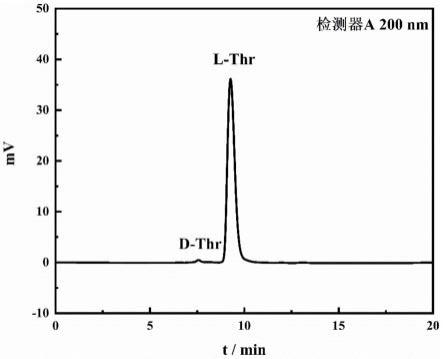
(2)重复实施例6中步骤(1)、(2)和(3),循环2次操作即可获得高纯度产品,产品的hplc色谱检测(方法同实施例4)的色谱图如图10所示,在保留时间10.04min处存在l-thr,l-thr的峰面积百分比为99.54;在保留时间7.00min处存在d-thr,d-thr的峰面积百分比为0.46,由此可得:l-thr对映体过量,ee值为99.08%。
[0081]
实施例8利用实施例2制备得到的手性离子液体拆分外消旋苏氨酸
[0082]
(1)配置10wt%[hmim]2[l-ma]与水的潜溶剂,以实现低粘度的手性溶剂体系。过量的外消旋苏氨酸溶解在10wt%[hmim]2[l-ma]的水溶液中,并在313.15k下搅拌12小时后,静置12小时,使其达到固液平衡;
[0083]
(2)取出313.15k下的饱和溶液,放入连有程序控温装置的313.15k的结晶器中,以350rpm的搅拌转速,0.1k/min的降温速率开始降温,析出产品后开始取样,每相隔5min进行一次取样,共取样15次,过滤获得固体产品。用高效液相色谱检测(方法同实施例4)15次样品的光学纯度,确定产品ee值最高的时间点;
[0084]
(3)重复步骤(1)和(2),在进行步骤(2)时只在产品ee值最高的时间点取样过滤获得产品;ee值最高时的hplc色谱图如图11所示,在保留时间9.34min处存在l-thr,l-thr的峰面积百分比为83.71;在保留时间7.10min处存在d-thr,d-thr的峰面积百分比为16.29,由此可得:l-thr对映体过量,ee值为67.42%;
[0085]
(4)将母液旋转蒸发除水,所得的混合物在冰浴的条件下用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去苏氨酸;收集滤液并旋转蒸发除去有机溶剂,放入353.15k的真空烘箱干燥48小时获得手性离子液体可再次使用。
[0086]
实施例9利用实施例2制备得到的手性离子液体拆分外消旋苏氨酸
[0087]
(1)配置300wt%[hmim]2[l-ma]与水的潜溶剂,以实现低粘度的手性溶剂体系。过量的外消旋苏氨酸溶解在30wt%[hmim]2[l-ma]的水溶液中,并在313.15k下搅拌24小时后,静置24小时,使其达到固液平衡;
[0088]
(2)取出313.15k下的饱和溶液,放入连有程序控温装置的313.15k的结晶器中,以以350rpm的搅拌转速,0.1k/min的降温速率开始降温,析出产品后开始取样,每相隔5min进行一次取样,共取样15次,过滤获得固体产品。用高效液相色谱检测(方法同实施例4)15次
样品的光学纯度,确定产品ee值最高的时间点;
[0089]
(3)重复步骤(1)和(2),在进行步骤(2)时只在产品ee值最高的时间点取样过滤获得产品,ee值最高时的hplc色谱图如图12所示,在保留时间9.34min处存在l-thr,l-thr的峰面积百分比为89.10;在保留时间7.11min处存在d-thr,d-thr的峰面积百分比为10.90,由此可得:l-thr对映体过量,ee值为78.2%;
[0090]
(4)将母液旋转蒸发除水,所得的混合物在冰浴的条件下用体积比9:1的乙腈和甲醇溶解并剧烈搅拌,过滤除去苏氨酸。收集滤液并旋转蒸发除去有机溶剂,放入353.15k的真空烘箱干燥48小时获得手性离子液体可再次使用。
[0091]
实施例10利用实施例2制备得到的手性离子液体拆分外消旋苏氨酸
[0092]
(1)将实施例9中步骤(3)中的固体产品过量溶于30wt%[hmim]2[l-ma]与水的潜溶剂,并在313.15k下搅拌24小时后,静置24小时,使其达到固液平衡;
[0093]
(2)重复实施例9中步骤(1)、(2)和(3),循环1次操作即可获得高ee值产品,产品的hplc色谱检测(方法同实施例4)的色谱图如图13所示,在保留时间9.27min处存在l-thr,l-thr的峰面积百分比为99.61;在保留时间7.09min处存在d-thr,d-thr的峰面积百分比为0.39,由此可得:l-thr对映体过量,ee值为99.22%。
[0094]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1