一种聚芳醚醚酮酮前驱体树脂及一种聚芳醚醚酮酮树脂的制备方法
1.本发明属于聚酰亚胺泡沫材料技术领域,涉及一种聚芳醚醚酮酮树脂前驱体、一种聚芳醚醚酮酮树脂及其制备方法,尤其涉及一种聚芳醚醚酮酮前驱体树脂及一种聚芳醚醚酮酮树脂的制备方法。
背景技术:
2.聚醚酮类树脂是20世纪70年代末研究开发成功的一类新型半晶态芳香族热塑性工程塑料,是全世界性能最高的热塑性材料之一,因其具有机械强度高、耐高温、耐冲击、阻燃、耐酸碱、耐水解、耐磨、耐疲劳、耐辐照及良好的电性能,在国防军工、航空航天、电子信息、能源、汽车、家电、医疗卫生等高新技术领域中取得了广泛的应用。
3.在英国专利bp1414421、美国专利us4320224、美国专利us4638044以及美国专利us4774314中报道了一系列经典结构的聚芳醚酮类树脂及其制备路线。在上述专利中得到的这些树脂分子主链均为刚性链且可以规整排列,故而导致相应树脂的溶解性极差。制备过程一般是用二苯砜做溶剂,碱催化,在高温条件下(一般250~350℃),脱水聚合。由于二苯砜水溶性差,一般要用水溶性有机溶剂萃取,然后水洗除盐和溶剂,获得纯化树脂。该过程流程长、溶剂消耗量大,生产成本高,环境污染和安全风险较大。还如专利cn106633034 a中所公开的,使用二氟二苯酮和苯甲胺反应制备席夫碱单体,后和对苯二酚,在n-甲基吡咯烷酮(nmp)中,用碳酸钾催化,甲苯共沸带水,然后聚合制得聚合物,然后酸催化,脱出苯胺,制得聚醚酮树脂。该方法碳酸钾使用量较大,使用易燃的甲苯做带水剂,反应时间长,生产成本依然较高。
4.同时,现有已开发制备聚芳醚醚酮酮技术的缺点主要有:1)聚合温度高,存在安全隐患;2)使用二苯砜,后处理过程繁琐,增加生产成本;3)带水法制备:耗时长、使用甲苯,增加成本;4)传统使用大颗粒催化剂,催化效果差,使用量多;5)高结晶聚芳醚醚酮酮难以溶解,分子量、分子量分布等不易表征,难以溶液加工成型。
5.因此,如何找到一种更为适宜的方式,解决现有的上述制备聚芳醚醚酮酮类树脂存在的问题,对进一步拓宽其在后续应用中的深度和广度具有重要意义,也是业内诸多研发人员亟待解决的问题之一。
技术实现要素:
6.有鉴于此,本发明要解决的技术问题在于提供一种聚芳醚醚酮酮树脂前驱体、一种聚芳醚醚酮酮树脂及其制备方法,特别是聚芳醚醚酮酮前驱体树脂以及相应的聚芳醚醚酮酮树脂的制备方法。本发明制备的该类前驱体树脂 (peekki)具有无定形聚集态、可溶于dmf等常规有机溶剂中、合成条件相对温和、分子量可通过gpc等方法测定。该类前驱体树脂经酸化处理后可生成相应的聚芳醚醚酮酮树脂(peekk)。因而解决了聚芳醚醚酮酮类树脂的低成本和安全合成、分子量测定、及溶液加工成型等问题;而且工艺简单、易于控制,有利
于实现工业化规模生产和应用。
7.本发明提供了一种聚芳醚醚酮酮树脂前驱体,具有如式(i)所示的结构:
[0008][0009]
其中,r1选自选自
[0010]
r2选自其中,r4为h、f、cl、br、ch3、 ch(ch3)2、c(ch3)3、苯基、no2、och3和so3h,m为1~5;
[0011]
r3选自自
[0012]
n为聚合度。
[0013]
优选的,所述n为25~400;
[0014]
所述前驱体为可溶性化合物;
[0015]
所述前驱体经过酸性水解后,得到聚芳醚醚酮酮树脂。
[0016]
本发明提供了一种聚芳醚醚酮酮树脂,具有如式(ii)所示的结构:
[0017][0018]
其中,r1选自选自
[0019]
r3选自自
[0020]
n为聚合度。
[0021]
本发明提供了一种聚芳醚醚酮酮树脂的制备方法,包括以下步骤:
[0022]
1)将二酮单体、一级胺、酸性化合物催化剂和有机溶剂混合,进行反应,得到席夫碱单体;
[0023]
2)在保护性气氛下,将上述步骤得到的席夫碱单体、二酚单体、碱性化合物催化剂和溶剂进行聚合反应,再加入封端剂继续反应后,得到聚芳醚醚酮酮树脂前驱体;
[0024]
3)将上述步骤得到的聚芳醚醚酮酮树脂前驱体在强酸溶液中进行水解反应后,得到聚芳醚醚酮酮树脂。
[0025]
优选的,所述二酮单体包括双卤双酮单体;
[0026]
所述二酮单体具有如式(iii)所示的结构:
[0027][0028]
其中,x为f、cl、no2;r1选自选自
[0029][0030]
所述一级胺具有如式(iv)所示的结构:
[0031]r2-nh2ꢀꢀꢀ
(iv);
[0032]
其中,r2选自其中,r4为h、f、cl、br、 ch3、ch(ch3)2、c(ch3)3、苯基、no2、och3和so3h,m为1~5。
[0033]
优选的,所述酸性化合物催化剂包括甲苯磺酸、分子筛和乙酸中的一种或多种;
[0034]
所述二酮单体和一级胺的摩尔比为1:(2~10);
[0035]
所述二酮单体和酸性化合物催化剂的摩尔比为1:(0.01~0.05);
[0036]
所述反应的温度为120~160℃;
[0037]
所述反应的时间为3~6h。
[0038]
优选的,所述二酚单体具有如式(v)所示的结构:
[0039]
ho-r
3-oh
ꢀꢀꢀ
(v);
[0040]
其中,r3选自选自
[0041]
所述碱性化合物催化剂包括碳酸钾、碳酸钠和碳酸铯中的一种或多种;
[0042]
所述碱性化合物催化剂的粒度为10~100μm;
[0043]
所述溶剂包括二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、1,3-二甲基-2
‑ꢀ
咪唑啉酮和n-甲基吡咯烷酮中的一种或多种;
[0044]
所述碱性化合物催化剂与双酚单体的摩尔比为(1.05~2):1;
[0045]
所述席夫碱单体与双酚单体的摩尔比为(0.95~1.05):1;
[0046]
所述溶剂的体积与席夫碱单体和双酚单体的总质量的比值为(1~2):1。
[0047]
优选的,所述聚合反应包括预热反应过程和反应过程;
[0048]
所述预热反应过程的温度为180~200℃;
[0049]
所述预热反应过程的时间为0.5~1h;
[0050]
所述反应过程的温度为210~230℃;
[0051]
所述反应过程的时间为1~10h;
[0052]
所述封端剂包括氯甲烷;
[0053]
所述继续反应的时间为15~60min。
[0054]
优选的,所述步骤2)还可以为以下步骤:
[0055]
2`)在保护性气氛下,将上述步骤得到的席夫碱单体、二酚单体、碱性化合物催化剂、带水剂和溶剂进行加热反应,蒸出带水剂后继续进行聚合反应,再加入封端剂继续反应后,得到聚芳醚醚酮酮树脂前驱体;
[0056]
所述带水剂包括环己烷、甲苯、二甲苯和氯苯中的一种或多种;
[0057]
所述加热反应的温度为120~180℃;
[0058]
所述加热反应的时间为2~6h;
[0059]
所述继续进行聚合反应的温度为160~200℃;
[0060]
所述继续进行聚合反应的时间为4~12h;
[0061]
所述封端剂包括氯甲烷;
[0062]
所述继续反应的时间为15~60min。
[0063]
优选的,所述强酸溶液中的强酸包括盐酸、硫酸、磷酸、氢溴酸、氢碘酸、甲烷磺酸和三氟乙酸中的一种或多种;
[0064]
所述强酸溶液中的溶剂包括水、甲醇、乙醇、nmp、乙酸和四氢呋喃中的一种或多种;
[0065]
所述水解反应的温度为60~100℃;
[0066]
所述水解反应的时间为2~24h。
[0067]
本发明提供了一种聚芳醚醚酮酮树脂前驱体,具有如式(i)所示的结构。与现有技术相比,本发明针对聚芳醚醚酮酮类树脂,其结晶度高、熔融温度高、难溶于常规有机溶剂,传统方法须在230℃以上、利用二苯砜等高沸点溶剂、在熔融状态下聚合生成。面临生产成本高、安全和环境污染风险高、聚合物分子量难以测量等问题。而且聚芳醚醚酮酮类树脂通常仅可通过熔融注塑加工成型,难以溶液加工,极大限制了其应用的缺陷。本发明特别设计了一种具有特定结构的聚芳醚醚酮酮(peekk)前驱体树脂,该类树脂具有无定形聚集态、可溶于n,n-二甲基甲酰胺(dmf)等常规有机溶剂、合成条件相对温和、分子量可通过gpc等方法测定,而且该类前驱体树脂经酸化处理后,即可生成相应的聚芳醚醚酮酮树脂。因而解决了聚芳醚醚酮酮类树脂的低成本和安全合成、分子量测定、及溶液加工成型等问题。
[0068]
本发明通过提高聚合物溶解度,解决高结晶聚醚酮类树脂聚合温度高,存在安全隐患问题;使用水溶性溶剂、避免使用带水剂,降低生产成本;使用小粒径催化剂,提高催化效率,减少催化剂使用量。本发明还能够通过表征聚芳醚醚酮酮前驱体的分子量和分子量分布,间接表征聚芳醚醚酮酮树脂分子量、分子量分布,解决高结晶聚芳醚酮类树脂分子量、分子量分布等不易表征问题。此外,本发明可以先将聚芳醚醚酮酮前驱体制成溶液,经浇铸、非溶剂诱导相转化等工艺成型后,经酸化处理、将聚芳醚醚酮酮前驱体转化成聚芳醚醚酮酮树脂。
[0069]
本发明还提供了聚芳醚醚酮酮树脂前驱体的制备方法,以双卤芳香双酮单体为原料,通过和胺类物质反应制备席夫碱单体,然后和双酚单体反应,制备高分子量的聚芳醚醚酮酮前驱体,在通过酸性水解,制备聚芳醚醚酮酮树脂。关键点在于(1)筛选出具有能和胺类物质生成席夫碱的双卤双酮单体; (2)聚芳醚醚酮酮前驱体可溶,因此可以使用带水和不带水两种途径,使用水溶性溶剂,在较低温度下进行制备;(3)通过采用平均粒度为10~30μm的无水碳酸钾作为碱催化剂。由于反应体系温度的提高,一方面,平均粒度更小的无水碳酸钾能够在高温下迅速与双酚单体充分接触并发生反应,另一方面,高温下缩聚反应生成的水能够加速逃离聚合溶剂体系,抑制了双卤单体发生水解等副反应,进而避免使用易燃、易挥发且有毒的有机分水剂(例如:甲苯、二甲苯或氯苯等)。本发明提供的制备方法,缩短了聚合反应时间,提高了聚合反应效率,能够制备高性能聚芳醚醚酮酮树脂,而且工艺简单、易于控制,有利于实现工业化规模生产和应用。
[0070]
实验结果表明,相较于聚醚酮传统聚合工艺,本发明所需聚合温度更低 (传统工艺为250~320℃,本方案为160~220℃),所需碱催化剂更少(传统工艺为1.5~2.1倍量,本发明可以低至1.05倍量),能耗消耗更少,使用水溶性溶剂更易去除,减少其他溶剂使用(传统工艺需使用丙酮类水溶性溶剂进行萃取)。相较于带水工艺,不带水工艺所需聚合时间更短(不带水工艺为 1~10h,带水工艺为4~12h),所用有机溶剂更少(不需要带水剂)。不带水工艺更适用于高温稳定的聚合单体,带水工艺则适用于高温欠稳定的聚合单体。本发明制备聚芳醚醚酮酮前聚体在dmf等传统溶剂具有良好的溶解度,可以用gpc进行分子量表征。
附图说明
[0071]
图1为本发明实施例1制备的席夫碱单体a1在cdcl3中1h nmr谱图;
[0072]
图2为本发明实施例2制备的席夫碱单体a2在d6-dmso中1h nmr谱图。
具体实施方式
[0073]
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为了进一步说明本发明的特征和优点,而不是对发明权利要求的限制。
[0074]
本发明所有原料,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知的常规方法制备的即可。
[0075]
本发明所有原料,对其纯度没有特别限制,本发明优选采用分析纯或聚芳醚醚酮酮树脂制备领域常规的纯度要求。
[0076]
本发明所有原料,其牌号和简称均属于本领域常规牌号和简称,每个牌号和简称在其相关用途的领域内均是清楚明确的,本领域技术人员根据牌号、简称以及相应的用途,能够从市售中购买得到或常规方法制备得到。
[0077]
聚芳醚醚酮酮类树脂:是在主链结构的重复单元中含有两个醚键和两个酮羰基的高分子聚合物,典型结构是一类半结晶性的高聚物。
[0078]
本发明提供了一种聚芳醚醚酮酮树脂前驱体,具有如式(i)所示的结构:
[0079][0080]
其中,r1选自选自
[0081]
r2选自其中,r4为h、f、cl、br、ch3、 ch(ch3)2、c(ch3)3、苯基、no2、och3和so3h,m为1~5;
[0082]
r3选自自
[0083]
n为聚合度。
[0084]
在本发明中,所述m为1~5,具体可以为1、2、3、4或5。
[0085]
在本发明中,所述n优选为25~400,更优选为50~350,更优选为100~300,更优选为150~250。
[0086]
在本发明中,所述前驱体优选为可溶性化合物。
[0087]
在本发明中,所述前驱体优选经过酸性水解后,得到聚芳醚醚酮酮树脂。
[0088]
本发明提供了一种聚芳醚醚酮酮树脂,具有如式(ii)所示的结构:
[0089][0090]
其中,r1选自选自
[0091]
r3选自自
[0092]
n为聚合度。
[0093]
在本发明中,所述n优选为25~400,更优选为50~350,更优选为100~300,更优选为150~250。
[0094]
本发明还提供了一种聚芳醚醚酮酮树脂的制备方法,包括以下步骤:
[0095]
1)将二酮单体、一级胺、酸性化合物催化剂和有机溶剂混合,进行反应,得到席夫碱单体;
[0096]
2)在保护性气氛下,将上述步骤得到的席夫碱单体、二酚单体、碱性化合物催化剂和溶剂进行聚合反应,再加入封端剂继续反应后,得到聚芳醚醚酮酮树脂前驱体;
[0097]
3)将上述步骤得到的聚芳醚醚酮酮树脂前驱体在强酸溶液中进行水解反应后,得到聚芳醚醚酮酮树脂。
[0098]
在本发明中,上述聚芳醚醚酮酮树脂的制备方法中包括聚芳醚醚酮酮前驱体树脂的制备方法,其中步骤1)和2)为聚芳醚醚酮酮前驱体树脂的制备方法。
[0099]
本发明首先将二酮单体、一级胺、酸性化合物催化剂和有机溶剂混合,进行反应,得到席夫碱单体。
[0100]
在本发明中,所述二酮单体优选包括双卤双酮单体。
[0101]
在本发明中,所述二酮单体优选具有如式(iii)所示的结构:
[0102][0103]
其中,x为f、cl、no2;r1选自选自
[0104]
所述一级胺具有如式(iv)所示的结构:
[0105]r2-nh2ꢀꢀꢀ
(iv);
[0106]
其中,r2选自其中,r4为h、f、cl、br、 ch3、ch(ch3)2、c(ch3)3、苯基、no2、och3和so3h,m优选为1~5,更优选为2~4。
[0107]
在本发明中,所述酸性化合物催化剂优选包括甲苯磺酸、分子筛和乙酸中的一种或多种,更优选为甲苯磺酸、分子筛或乙酸。
[0108]
在本发明中,所述二酮单体和一级胺的摩尔比优选为1:(2~10),更优选为1:(3.5~8.5),更优选为1:(5~7)。
[0109]
在本发明中,所述二酮单体和酸性化合物催化剂的摩尔比优选为1: (0.01~0.05),更优选为1:(0.015~0.045),更优选为1:(0.02~0.04),更优选为1:(0.025~0.035)。
[0110]
在本发明中,所述反应的温度优选为120~160℃,更优选为125~155℃,更优选为
130~150℃,更优选为135~145℃。
[0111]
在本发明中,所述反应的时间优选为3~6h,更优选为3.5~5.5h,更优选为4~5h。
[0112]
本发明再在保护性气氛下,将上述步骤得到的席夫碱单体、二酚单体、碱性化合物催化剂和溶剂进行聚合反应,再加入封端剂继续反应后,得到聚芳醚醚酮酮树脂前驱体。
[0113]
在本发明中,所述二酚单体优选具有如式(v)所示的结构:
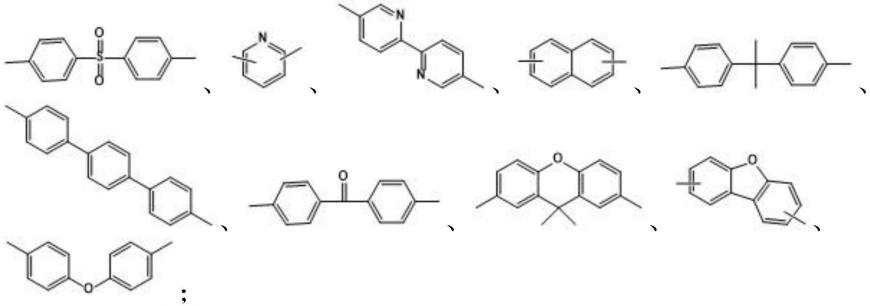
[0114]
ho-r
3-oh
ꢀꢀꢀ
(v);
[0115]
其中,r3选自选自
[0116]
在本发明中,所述碱性化合物催化剂优选包括碳酸钾、碳酸钠和碳酸铯中的一种或多种,更优选为碳酸钾、碳酸钠或碳酸铯。
[0117]
在本发明中,所述碱性化合物催化剂的粒度优选为10~100μm,更优选为 30~80μm,更优选为50~60μm。
[0118]
在本发明中,所述溶剂优选包括二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、1,3-二甲基-2-咪唑啉酮和n-甲基吡咯烷酮中的一种或多种,更优选为二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、1,3-二甲基-2-咪唑啉酮或n-甲基吡咯烷酮。
[0119]
在本发明中,所述碱性化合物催化剂与双酚单体的摩尔比优选为(1.05~2):1,更优选为(1.2~1.8):1,更优选为(1.4~1.6):1。
[0120]
在本发明中,所述席夫碱单体与双酚单体的摩尔比为(0.95~1.05):1,更优选为(0.97~1.03):1,更优选为(0.99~1.01):1。
[0121]
在本发明中,所述溶剂的体积与席夫碱单体和双酚单体的总质量的比值优选为(1~2):1,更优选为(1.2~1.8):1,更优选为(1.4~1.6):1。
[0122]
在本发明中,所述聚合反应优选包括预热反应过程和反应过程。
[0123]
在本发明中,所述预热反应过程的温度优选为180~200℃,更优选为 184~196℃,更优选为188~192℃。
[0124]
在本发明中,所述预热反应过程的时间优选为0.5~1h,更优选为0.6~0.9h,更优选为0.7~0.8h.
[0125]
在本发明中,所述反应过程的温度优选为210~230℃,更优选为 214~226℃,更优选为218~222℃。
[0126]
在本发明中,所述反应过程的时间优选为1~10h,更优选为3~8h,更优选为5~6h。
[0127]
在本发明中,所述封端剂优选包括氯甲烷。
[0128]
在本发明中,所述继续反应的时间优选为15~60min,更优选为25~50min,更优选为35~40min。
[0129]
在本发明中,所述步骤2)还优选可以为以下步骤:
[0130]
2`)在保护性气氛下,将上述步骤得到的席夫碱单体、二酚单体、碱性化合物催化剂、带水剂和溶剂进行加热反应,蒸出带水剂后继续进行聚合反应,再加入封端剂继续反应后,得到聚芳醚醚酮酮树脂前驱体。
[0131]
在本发明中,所述带水剂优选包括环己烷、甲苯、二甲苯和氯苯中的一种或多种,更优选为环己烷、甲苯、二甲苯或氯苯。
[0132]
在本发明中,所述加热反应的温度优选为120~180℃,更优选为 130~170℃,更优选为140~160℃。
[0133]
在本发明中,所述继续进行聚合反应的时间优选为4~12h,更优选为 6~10h,更优选为7~8h。
[0134]
在本发明中,所述封端剂优选包括氯甲烷。
[0135]
在本发明中,所述继续反应的时间优选为15~60min,更优选为25~50min,更优选为35~40min。
[0136]
本发明最后将上述步骤得到的聚芳醚醚酮酮树脂前驱体在强酸溶液中进行水解反应后,得到聚芳醚醚酮酮树脂。
[0137]
在本发明中,所述强酸溶液中的强酸优选包括盐酸、硫酸、磷酸、氢溴酸、氢碘酸、甲烷磺酸和三氟乙酸中的一种或多种,更优选为盐酸、硫酸、磷酸、氢溴酸、氢碘酸、甲烷磺酸或三氟乙酸。
[0138]
在本发明中,所述强酸溶液中的溶剂优选包括水、甲醇、乙醇、nmp、乙酸和四氢呋喃中的一种或多种,更优选为水、甲醇、乙醇、nmp、乙酸或四氢呋喃。
[0139]
在本发明中,所述水解反应的温度优选为60~100℃,更优选为68~92℃,更优选为76~84℃。
[0140]
在本发明中,所述水解反应的时间优选为2~24h,更优选为7~19h,更优选为12~14h。
[0141]
本发明提供的新的聚芳醚醚酮酮制备方法,通过制备席夫碱单体,然后和二酚单体,在选取特定粒径大小无水碳酸钾的作为催化剂和水溶性物质作溶剂条件下,使用或不使用带水剂两种途径,均可通过脱水缩聚获得高分子量的聚芳醚醚酮酮前驱体,在通过酸性水解,获得聚芳醚醚酮酮。该制备方法耗时短,反应温度低,能够避免使用易燃、易挥发且有毒的有机分水剂(甲苯、二甲苯或氯苯等)。
[0142]
本发明为完整和细化整体技术方案,更好的保证聚芳醚醚酮酮前驱体树脂以及聚芳醚醚酮酮树脂的性能和结构,上述聚芳醚醚酮酮制备方法具体可以为以下步骤:
[0143]
聚芳醚醚酮酮(peekk)聚合物的制备方法,包括如下步骤:
[0144]
(1)对二酮单体进行修饰制备席夫碱单体;
[0145]
(2)将席夫碱单体和二酚单体反应,使用弱碱作催化剂和水溶性物质作溶剂,经脱水聚合获得聚合物,然后水解获得聚芳醚醚酮酮。
[0146]
席夫碱单体的制备:二酮单体与有机一级胺在有机溶剂,在酸(对甲苯磺酸、分子筛、醋酸等)作为催化剂,反应3~6h,至不在有水生成,蒸干有机溶剂,沉于甲醇或乙醇,过
滤,洗涤数次,得席夫碱单体粗产品。重结晶得到高纯度席夫碱单体。
[0147]
席夫碱单体制备反应方程式如下:
[0148][0149]
具体的,本发明优选苯胺和对甲苯胺对二酮单体进行修饰。
[0150]
具体的,酸可以选用对甲苯磺酸、分子筛、乙酸等,优先选用对甲苯磺酸。
[0151]
具体的,二酮单体、胺和酸用量比(摩尔比)为1:(2~10):(1%~5%)。
[0152]
具体的,用乙烷/丙酮(1:1)重结晶,得到高纯度席夫碱单体。
[0153]
聚芳醚醚酮酮(peekk)聚合物制备:
[0154][0155]
聚芳醚醚酮酮的制备方法(一)-步骤2`):向配有分水器的反应釜中加入溶剂、带水剂和单体,搅拌条件加入弱碱。氮气保护条件下加热至 120~180℃,反应一段时间,待出水量平稳后,蒸出带水剂。升温至160~200℃,继续反应4~12h。降温至120℃,通入氯甲烷封端,反应0.5h。冷却室温,加入溶剂稀释,搅拌均匀,过滤,沉入水中,得聚合物树脂,多次洗涤,离心过滤,干燥备用。上述聚合物加入强酸溶液中,60~100℃时,恒温反应2~24 小时,反应生成物为聚芳醚醚酮酮树脂粗品。通过纯化,得到聚芳醚醚酮酮树脂。
[0156]
聚芳醚醚酮酮的制备方法(二)-步骤2):向配有分水器的反应釜中加入溶剂和单
体,搅拌条件加入弱碱。氮气保护条件下加热至180~200℃,反应一段时间,待出水量平稳后。升温至210~230℃,继续反应1~10h。降温至120℃,通入氯甲烷封端,反应0.5h。冷却室温,加入溶剂稀释,搅拌均匀,过滤,沉入水中,得聚合物树脂,多次洗涤,离心过滤,干燥备用。上述聚合物加入强酸溶液中,60~100℃时,恒温反应2~24小时,反应生成物为聚芳醚醚酮酮树脂粗品。通过纯化,得到聚芳醚醚酮酮树脂。
[0157]
具体的,带水剂可选环己烷、甲苯、二甲苯、氯苯。
[0158]
具体的,溶剂可选二甲基甲酰胺(dmf)、二甲基乙酰胺(dmac)、二甲基亚砜(dmso)、1,3-二甲基-2-咪唑啉酮(dmi)和n-甲基吡咯烷酮(nmp),优选nmp和dmi。
[0159]
具体的,溶剂与单体的体积质量比可为1~2:1,优选1:1
[0160]
具体的,弱碱可选碳酸钾、碳酸钠、碳酸铯,优选碳酸钾。
[0161]
具体的,弱碱平均粒径可为10~100μm,优选10~50μm,更优为10~30μm。
[0162]
具体的,弱碱与双酚单体用量比(摩尔比)为1.05~2:1,优选1.05~1.3,更优1.05~1.1:1。
[0163]
具体的,席夫碱单体与双酚单体用量比(摩尔比)为0.95~1.05:1,优选 0.99~1.01:1。
[0164]
具体的,对于制备方法(一):带水温度可为120~180℃,聚合温度为 160~200℃。
[0165]
具体的,对于制备方法(二):预热温度可为180~200℃,优选200℃。聚合温度可为200~230℃,优选220℃。
[0166]
具体的,强酸可选盐酸、硫酸、磷酸、氢溴酸、氢碘酸、甲烷磺酸、三氟乙酸等一种或几种,优选盐酸。可选水溶液、甲醇溶液、乙醇溶液、nmp 溶液、乙酸溶液、四氢呋喃溶液或其中几种的混合溶液,优选乙醇溶液。
[0167]
具体的,水解温度可为60~100℃,优选80℃。
[0168]
具体的,本发明采用具有高温稳定的双卤双酮单体为原料,其结构特征是:酮羰基可以和胺类物质反应。更为具体结构如下所示:
[0169][0170]
其中x为f、cl、no2,r1为
[0171]
具体的,本发明对有机胺结构没有特殊限定,一般为有机一级胺,更为具体结构如下所示:r
2-nh2。
[0172]
其中r2为其中r4为h、f、cl、br、ch3、 ch(ch3)2、c(ch3)3、苯基、no2、och3和so3h,n为1~5。
[0173]
具体的,本发明对双酚单体结构没有特殊限定,更为具体结构如下所示:
[0174]
ho-r
3-oh。
[0175]
其中,r3为为
[0176]
具体的,本发明的聚芳醚醚酮酮前驱体可以使用带水和不带水两种途径进行制备:
[0177]
带水途径:带水剂一般选用甲苯、二甲苯、氯苯,溶剂可选dmac、dmf、 dmso、nmp,聚合温度为180~200℃,聚合时间为4~12h。
[0178]
不带水途径:溶剂首选nmp、dmi,聚合温度为200~230℃,聚合时间为1~10h。
[0179]
具体的,本发明未处理碳酸钾用量一般为双酚单体的1.5~2.1倍量。以平均粒度10~100μm的无水碳酸钾作为碱催化剂,用量一般为双酚单体的 1.05~1.3倍量。更为具体的是:选用平均粒度10~30μm的无水碳酸钾其摩尔当量数可以降至双酚单体1.05倍。
[0180]
本发明通过制备席夫碱单体,可以提高聚合物溶解度,解决随着聚合物分子量增加,聚合物析出的问题,从而制备高分子量聚合物,且可以表征分子量、分子量分布。(2)由于聚合物可溶,可以选用较低沸点水溶性溶剂,后期可以通过水洗,去除溶剂和水溶性物质。(3)选用较小粒径催化剂可以加速缩聚反应生成的水逃离聚合溶剂体系,不仅抑制了双卤单体的水解等副反应的发生,而且还避免使用易燃、易挥发且有毒的有机分水剂,例如:
甲苯、二甲苯或氯苯等。该制备方法缩短了聚合反应时间,提高了聚合反应效率,制备高性能聚芳醚醚酮酮树脂材料。
[0181]
本发明从底物的适用范围角度上看,该方法适用于制备高结晶度聚醚酮类树脂。双卤单体的结构特征是:酮羰基可以胺类物质反应生成席夫碱结构;双酚单体结构没有特殊限定。底物范围具有广泛的适用性。从聚合反应效率角度上看,通过提高聚合反应温度,缩短了该类聚合反应的时间,一般2~5 小时即可完成,提高了聚合反应效率。从聚合反应效果角度上看,选用平均粒度为10~30μm的无水碳酸钾其摩尔当量数可以降至双酚单体1.05倍,而传统缩聚方法通常需要1.50~2.00当量的无水碳酸钾。该方法大大降低了无水碳酸钾使用量。从环境保护角度上看,该方法可以避免使用易燃、易挥发且有毒的有机分水剂(甲苯、二甲苯或氯苯等),有效地降低了环境污染。
[0182]
本发明上述内容提供了一种聚芳醚醚酮酮前驱体树脂及其制备方法、一种聚芳醚醚酮酮树脂及其制备方法。本发明特别设计的具有特定结构的聚芳醚醚酮酮(peekk)前驱体树脂,该类树脂具有无定形聚集态、可溶于n, n-二甲基甲酰胺(dmf)等常规有机溶剂、合成条件相对温和、分子量可通过gpc等方法测定,而且该类前驱体树脂经酸化处理后,即可生成相应的聚芳醚醚酮酮树脂。因而解决了聚芳醚醚酮酮类树脂的低成本和安全合成、分子量测定、及溶液加工成型等问题。
[0183]
本发明通过提高聚合物溶解度,解决高结晶聚醚酮类树脂聚合温度高,存在安全隐患问题;使用水溶性溶剂、避免使用带水剂,降低生产成本;使用小粒径催化剂,提高催化效率,减少催化剂使用量。本发明还能够通过表征聚芳醚醚酮酮前驱体的分子量和分子量分布,间接表征聚芳醚醚酮酮树脂分子量、分子量分布,解决高结晶聚芳醚酮类树脂分子量、分子量分布等不易表征问题。此外,本发明可以先将聚芳醚醚酮酮前驱体制成溶液,经浇铸、非溶剂诱导相转化等工艺成型后,经酸化处理、将聚芳醚醚酮酮前驱体转化成聚芳醚醚酮酮树脂。
[0184]
本发明还提供了聚芳醚醚酮酮树脂前驱体的制备方法,以双卤芳香双酮单体为原料,通过和胺类物质反应制备席夫碱单体,然后和双酚单体反应,制备高分子量的聚芳醚醚酮酮前驱体,在通过酸性水解,制备聚芳醚醚酮酮树脂。关键点在于(1)筛选出具有能和胺类物质生成席夫碱的双卤双酮单体; (2)聚芳醚醚酮酮前驱体可溶,因此可以使用带水和不带水两种途径,使用水溶性溶剂,在较低温度下进行制备;(3)通过采用平均粒度为10~30μm的无水碳酸钾作为碱催化剂。由于反应体系温度的提高,一方面,平均粒度更小的无水碳酸钾能够在高温下迅速与双酚单体充分接触并发生反应,另一方面,高温下缩聚反应生成的水能够加速逃离聚合溶剂体系,抑制了双卤单体发生水解等副反应,进而避免使用易燃、易挥发且有毒的有机分水剂(例如:甲苯、二甲苯或氯苯等)。本发明提供的制备方法,缩短了聚合反应时间,提高了聚合反应效率,能够制备高性能聚芳醚醚酮酮树脂,而且工艺简单、易于控制,有利于实现工业化规模生产和应用。
[0185]
实验结果表明,相较于聚醚酮传统聚合工艺,本发明所需聚合温度更低 (传统工艺为250~320℃,本方案为160~220℃),所需碱催化剂更少(传统工艺为1.5~2.1倍量,本发明可以低至1.05倍量),能耗消耗更少,使用水溶性溶剂更易去除,减少其他溶剂使用(传统工艺需使用丙酮类水溶性溶剂进行萃取)。相较于带水工艺,不带水工艺所需聚合时间更短(不带水工艺为 1~10h,带水工艺为4~12h),所用有机溶剂更少(不需要带水剂)。不带
水工艺更适用于高温稳定的聚合单体,带水工艺则适用于高温欠稳定的聚合单体。本发明制备聚芳醚醚酮酮前聚体在dmf等传统溶剂具有良好的溶解度,可以用gpc进行分子量表征。
[0186]
为了进一步说明本发明,以下结合实施例对本发明提供的一种聚芳醚醚酮酮树脂前驱体、一种聚芳醚醚酮酮树脂及其制备方法进行详细描述,但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
[0187]
单体制备
[0188]
实施案例1
[0189]
向配有分水器的反应釜中加入500ml甲苯,然后将二氟三苯二酮a(1.00 mol,50g)、对甲苯磺酸(4g)和苯胺1(1.00mol,60g)溶于于溶剂中,氮气保护条件下加热至180℃,反应5.0小时。蒸出甲苯,甲醇洗涤,干燥得到粗产品。用丙酮/己烷(1:1)重结晶的高纯度席夫碱单体a1。产品:26g,产率:55%。
[0190]
对本发明实施例1制备的席夫碱单体a1进行表征。
[0191]
参见图1,图1为本发明实施例1制备的席夫碱单体a1在cdcl3中1hnmr谱图。
[0192]
实施案例2
[0193]
向配有分水器的反应釜中加入500ml甲苯,然后将二氟三苯二酮a(0.15 mol,50g)、对甲苯磺酸(4g)和对甲苯胺2(0.37mol,40g)溶于于溶剂中,氮气保护条件下加热至180℃,反应5.0小时。蒸出甲苯,甲醇洗涤,干燥得到粗产品。用丙酮/己烷(1:1)重结晶的高纯度席夫碱单体a2。产品: 60g,产率:78%。
[0194]
对本发明实施例2制备的席夫碱单体a2进行表征。
[0195]
参见图2,图2为本发明实施例2制备的席夫碱单体a2在d6-dmso中1h nmr谱图。
[0196]
聚合反应
[0197]
实施案例3
[0198]
向配有分水器的反应釜中加入50mlnmp溶剂,然后将单体a1(0.042 mol,20g)和对苯二酚(c1)(0.042mol,4.66g)悬浮于溶剂中,搅拌条件加入平均粒径为10-100μm无水碳酸钾(0.055mol,7.6g)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应4 小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450mlnmp搅拌均匀,过滤,沉入水中,多次洗涤,200℃真空干燥得聚合物 peekki-1。
[0199]
实施案例4
[0200]
向配有分水器的反应釜中加入50ml nmp溶剂,然后将单体a2(0.040 mol,20g)和对苯二酚(c1)(0.040mol,4.40g)悬浮于溶剂中,搅拌条件加入平均粒径为10-100μm无水碳酸钾(0.052mol,7.2g)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应4 小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450mlnmp搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得peekki-2。
[0201]
实施案例5
[0202]
向配有分水器的反应釜中加入56ml nmp溶剂,然后将单体a1(0.042 mol,20g)和联苯二酚(c2)(0.042mol,7.84g)悬浮于溶剂中,搅拌条件加入平均粒径为10-100μm无水碳
酸钾(0.055mol,7.6g)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应4 小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450mlnmp搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物 peekki-3。
[0203]
实施案例6
[0204]
向配有分水器的反应釜中加入56ml nmp溶剂,然后将单体a2(0.040 mol,20g)和联苯二酚(c2)(0.040mol,7.39g)悬浮于溶剂中,搅拌条件加入平均粒径为10~100μm无水碳酸钾(0.052mol,7.2g)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应 4小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450 ml nmp搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物 peekki-4-1。
[0205]
实施案例7
[0206]
向配有分水器的反应釜中加入28ml nmp溶剂,然后将单体a2(0.040 mol,20g)和联苯二酚(c2)(0.040mol,7.39g)悬浮于溶剂中,搅拌条件加入平均粒径为10μm、1.3~1.05当量无水碳酸钾。氮气保护条件下加热至 200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应至要求粘度。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450mlnmp搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物 peekki-4-2。
[0207]
实施案例8
[0208]
向配有分水器的反应釜中加入28ml dmi溶剂,然后将单体a2(0.040 mol,20g)和联苯二酚(c2)(0.040mol,7.39g)悬浮于溶剂中,搅拌条件加入平均粒径为10μm无水碳酸钾(0.044mol,6.1g)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应3小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450mldmi搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物 peekki-4-3。
[0209]
实施案例9
[0210]
向配有分水器的反应釜中加入56ml nmp溶剂,然后将单体a2(0.040 mol,20g)和联苯二酚(c2)(0.040mol,7.39g)悬浮于溶剂中,甲苯56ml,搅拌条件下无水碳酸钾。氮气保护条件下加热至160℃,反应1~3小时,待出水量平稳后。升温至180℃,继续反应5小时。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450ml dmi搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物peekki-4-4。
[0211]
实施案例10
[0212]
在氮气保护条件下,将聚合物peekki(聚芳醚醚酮酮前驱体)20g粉料投入100ml 1m hcl的乙醇溶液中,80℃,恒温反应24h,然后80℃水洗3次,干燥得到peekk树脂(聚芳醚醚酮酮)。
[0213]
对比实施案例1
[0214]
向配有分水器的反应釜中加入28ml nmp溶剂,然后将单体a2(0.040 mol,20g)和联苯二酚(c2)(0.040mol,7.39g)悬浮于溶剂中,搅拌条件加入无水碳酸钾(0.044mol,6.1g,商业购买未处理)。氮气保护条件下加热至200℃,反应1小时,待出水量平稳后。升温至230℃,继续反应10h。降温至120℃,通入氯甲烷封端,反应0.5h。降至室温后,加入450ml nmp 搅拌均匀,过滤,沉入水中,多次洗涤,140℃真空干燥得聚合物peekki-4-5。
[0215]
对比实施案例2
[0216]
向配有分水器的反应釜中加入二氟三苯二酮(0.05mol,16.16g)、联苯二酚
(0.05mol,9.26g)及溶剂二苯砜(21.8g),通氮气并加热升温至180℃,加入无水碳酸钾(0.1mol,13.8g),升温至200℃保温1h,然后再升温至 250℃,反应15min,最终升温至320℃,反应2.5h。冷却至室温,聚合物粉碎后,用丙酮萃取,以除去二苯砜。用水洗涤,以除去聚合物中的无机盐。 140℃真空干燥得聚合物peekk。
[0217]
单体a、peekk-1,2和peeki-(1~4)的化学结构式
[0218][0219][0220]
参见表1,表1为本发明实施例制备的peekki的聚合条件和聚合物分子量测定。
[0221]
表1
[0222][0223]
参见表2,表2为本发明制备的peekki和peekk的溶解性对照表。
[0224]
表2
[0225][0226][0227]
其中,
“‑”
代表不溶,“+”代表可溶。
[0228]
以上对本发明提供的一种聚芳醚醚酮酮前驱体树脂及一种聚芳醚醚酮酮树脂的制备方法进行了详细的介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有不是不同于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1