一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法与流程
1.本发明属于有机合成技术领域,具体涉及一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法。
背景技术:
2.络氨酸激酶是一组催化蛋白质络氨酸残基磷酸化的酶,在细胞内的信号转导中起着重要的作用,它参与正常细胞的调节、信号传递和发育,也与肿瘤细胞的增殖、分化、迁移和凋亡密切相关。许多受体络氨酸激酶都与肿瘤的形成相关,根据其细胞外区域结构的不同可分为表皮生长因子受体(egfr)、血小板衍化生长因子受体(pdgfr)、血管内皮细胞生长因子受体(vegfr)、成纤维细胞生长因子受体(fgfr)等。
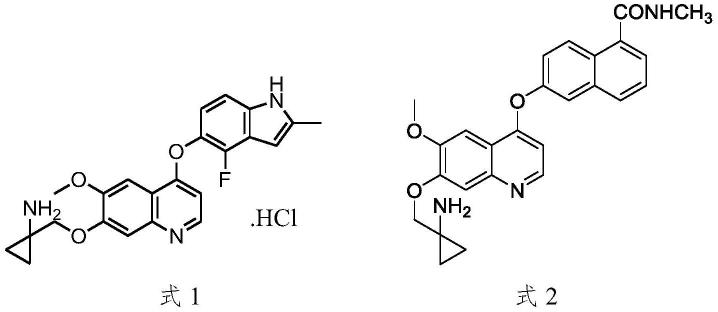
3.盐酸安罗替尼是一种新型小分子多靶点酪氨酸激酶抑制剂,能有效抑制vegfr、pdgfr、fgfr、c-kit等激酶,具有抗肿瘤血管生成和抑制肿瘤生长的作用,目前已上市。德立替尼是同时靶向成纤维细胞生长因子受体1-2(fgfr1-2)和内皮生长因子受体1-3(vegfr1-3)的受体酪氨酸激酶抑制剂,在特异治疗fgfr1-2依赖型肿瘤的同时,还具有广泛的抑制肿瘤新生血管生成的作用。其中,盐酸安罗替尼的结构如下式1所示;德立替尼的结构如下式2所示。
[0004][0005]
4-氯-6-甲氧基-7-苄氧基喹啉可作为上述制备抗肿瘤药物盐酸安罗替尼和德立替尼药物分子合成的关键中间体。
[0006]
关于中间体4-氯-6-甲氧基-7-苄氧基喹啉的制备方法有如下报导,现有方法一:forsyth,tim-otly,patrick.mac,morrison,b.等人在专利wo2006108059a1中公布:用4-苄氧基-3-甲氧基苯乙酮为原料,以硫酸为溶剂,与硝酸在低温0℃进行硝化制备得1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮,再用铁粉和醋酸胺作用下还原,得到(e)-1-(4-(苄氧基)-5-甲氧基-2-氨基苯基)-3-(二甲基氨基)-2-丙烯-1-酮,再在乙二醇二甲醚和甲醇钠作用下环合得到7-苄氧基-6-甲氧基-1h-喹啉-4-酮;再经氯代可得4-氯-6-甲氧基-7-苄氧基喹啉,具体反应路线如下式3所示:
[0007][0008]
该制备工艺路线长,该反应采取强碱环合的方法,需要无水条件,工业化成本高;采用铁粉还原硝基的方法,会生成大量的工业废渣,会带来严重的环境污染问题,工业化制备成本高。
[0009]
现有方法二:专利acta chimica hungarica,112(z),241-7;1983;u.s.20100239576;wo2005121125a1,报道了类似化合物的合成方法,该方法以3,4-二甲氧基苯胺为起始原料,与乙氧亚甲基丙二酸酯经亲核取代反应制备得化合物,在二苯醚-联苯中经250℃加热反应数h,得到4-羟基喹啉-3-甲酸乙酯化合物,再经过加热脱羧制备得4-羟基喹啉化合物,最后经三氯化磷氯代得到4-氯-6,7-二甲氧基喹啉。或以3,4-二甲氧基苯胺与乙氧亚甲基米氏酸经亲核加成制备得化合物,在二苯醚-联苯中经250℃加热反应数h,得到4-羟基喹啉,最后经三氯氧磷氯代可得4-氯-6,7-二甲氧基喹啉,其具体合成路线如下式4所示:
[0010][0011]
该制备路线需要采取高温环合方法,由于反应温度很高,会生成难溶性杂质,造成产品难以纯化,收率较低。另外采用二苯醚-联苯为溶剂,用量较大(约为原料的20倍质量)、沸点为260℃,难以回收;二苯醚-联苯价格相对较高且刺激性较大,会对操作人员和环境有较大危害。因此,高温环合的方法,对于放大生成不利。
[0012]
现有方法三:上海工程技术大学的朱春平等人在中国专利申请cn106008336a中,报道一种4-氯-6,7-二甲氧基喹啉的制备方法,以3,4-二甲氧基苯乙酮为原料,经硝酸硝化制备得到2-硝基-4,5-二甲氧基苯乙酮,再与n,n-二甲基甲酰胺二甲基缩醛缩合,经催化氢
化高温高压的情况发生还原环合制备得4-羟基-6,7-二甲氧基喹啉;经氯代得到4-氯-6,7-二甲氧基喹啉,其具体合成路线如下式5所示:
[0013][0014]
该方法需经过氢化高温高压的条件进行还原环合的反应条件,通过高温后产生难易去除的杂质,且对反应设备要求较高,属于高危工艺。
[0015]
基于现有技术方法存在的缺陷,本发明提出一种新的制备4-氯-6-甲氧基-7-苄氧基喹啉的方法,以提高4-氯-6-甲氧基-7-苄氧基喹啉的生产效率。
技术实现要素:
[0016]
本发明的目的在于,克服现有技术中存在的缺陷,提供一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法,以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮为起始原料,经过烯胺化、还原环合、氯代反应制备得4-氯-6-甲氧基-7-苄氧基喹啉,反应原料易得,不需高温高压,常压即可操作,反应条件温和,工艺简洁,操作简便,收率高,成本低,适合工业化放大生产。
[0017]
为实现上述目的,本发明的技术方案是设计一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法,包括如下步骤:
[0018]
s1:1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮与n,n-二甲基甲酰胺二甲缩醛缩合制备制得到(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮;
[0019]
s2:(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮经强还原环合剂还原反应后分子发生还原环合制备得到7-苄氧基-6-甲氧基-1h-喹啉-4-酮;
[0020]
s3:7-苄氧基-6-甲氧基-1h-喹啉-4-酮经氯代反应得到4-氯-6-甲氧基-7-苄氧基喹啉。
[0021]
优选的技术方案是,所述的步骤s1具体操作为:将1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮与第一溶剂混合,并加入n,n-二甲基甲酰胺二甲缩醛,于50-140℃下反应1-18h。
[0022]
进一步优选的技术方案还有,所述的步骤s1中,第一溶剂为甲苯、n,n-二甲基甲酰胺、乙二醇二甲醚、四氢呋喃中的一种,1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮与第一溶剂的质量投料比为1:0.1-3,1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮与n,n-二甲基甲酰胺二甲缩醛的摩尔投料比为1:1-4,反应温度为80-125℃,反应时间为2-8h。
[0023]
进一步优选的技术方案还有,所述的步骤s1中,1-(4-(苄氧基)-5-甲氧基-2-硝基
苯基)乙酮与第一溶剂的质量投料比为1:1-2,1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮与n,n-二甲基甲酰胺二甲缩醛的摩尔投料比为1:1.2-3。
[0024]
优选的技术方案还有,所述的步骤s2具体操作为:将(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮与第二溶剂混合,加入强还原环合剂搅拌均匀,在10-100℃下反应1-24h。
[0025]
进一步优选的技术方案还有,所述的步骤s2中,第二溶剂为无水乙醇、甲醇、异丙醇、乙醇/水混合溶液、甲醇/水混合溶液、异丙醇/水混合溶液,(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮与第二溶剂的质量投料比例为1:2~10,强还原环合剂为双氧水、连二亚硫酸钠、三乙酰氧基硼氢化钠、硼氢化钠、四氢铝锂、焦亚硫酸钠、二氧化硫脲、硫脲、n,n-二甲基甲酰胺中的一种,(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮与强还原环合剂的摩尔投料比为1:1-5,反应温度为20-90℃,反应时间为1.5-15h。
[0026]
进一步优选的技术方案还有,所述的步骤s2中,(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮与第二溶剂的质量投料比例为1:3,强还原环合剂为双氧水、连二亚硫酸钠、二氧化硫脲、硼氢化钠中的一种,(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮与强还原环合剂的摩尔投料比为1:3。
[0027]
优选的技术方案还有,所述的步骤s3具体操作为,将7-苄氧基-6-甲氧基-1h-喹啉-4-酮与第三溶剂混合,加入氯代试剂搅拌均匀,再加入缚酸剂,于30-105℃反应1-12h,将反应液淬灭,得粗品,重结晶纯化,得到4-氯-6-甲氧基-7-苄氧基喹啉。
[0028]
进一步优选的技术方案还有,所述的步骤s3中,氯代试剂为氯化亚砜、三光气、三氯化磷、三氯氧磷、五氯化磷中的一种,缚酸剂为三乙胺、二乙胺、n,n-二异丙基乙胺、三乙烯二胺或吡啶中的一种,第三溶剂为二氯甲烷、石油醚、正己烷、甲苯、1,2-二氯乙烷、氯仿、上述氯代试剂中的一种,重结晶溶剂为二氯甲烷、1,2-二氯乙烷、氯仿、甲苯、乙酸乙酯、甲醇、乙醇、丙酮、乙腈中的一种或几种,7-苄氧基-6-甲氧基-1h-喹啉-4-酮、氯代试剂、第三溶剂、缚酸剂的摩尔投料比为1:0.5-10:3:1-5,反应温度为40-100℃,反应时间为1-5h。
[0029]
进一步优选的技术方案还有,所述的步骤s3中,7-苄氧基-6-甲氧基-1h-喹啉-4-酮与缚酸剂的摩尔投料比为1:2.5,氯代试剂为三氯化磷、三氯氧磷中的一种。
[0030]
本发明的优点和有益效果在于:
[0031]
1、本发明的一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法,以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮为起始原料,经过烯胺化、还原环合、氯代反应制备得4-氯-6-甲氧基-7-苄氧基喹啉,反应原料易得,不需高温高压,常压即可操作,反应条件温和,工艺简洁,操作简便,收率高,成本低,适合工业化放大生产。
[0032]
2、本发明的一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法,在制备7-苄氧基-6-甲氧基-1h-喹啉-4-酮中,采用强还原环合剂二步一锅法:由原硝基还原为氨基后再经过环合试剂进行环合得到目标7-苄氧基-6-甲氧基-1h-喹啉-4-酮,不需经高温高压和通入氢气反应等高危工艺操作,即可制备得到7-苄氧基-6-甲氧基-1h-喹啉-4-酮,显著提高操作安全性。
附图说明
[0033]
图1是本发明一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法的合成路线图;
[0034]
图2是实施例1中制备的4-氯-6-甲氧基-7-苄氧基喹啉的高效液相色谱图;
[0035]
图3是实施例1中制备的4-氯-6-甲氧基-7-苄氧基喹啉的核磁图谱;
[0036]
图4是实施例1中制备的4-氯-6-甲氧基-7-苄氧基喹啉的碳谱图谱。
具体实施方式
[0037]
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
[0038]
实施例1
[0039]
采用本发明方法制备4-氯-6-甲氧基-7-苄氧基喹啉,合成路线参见附图1,包括如下操作步骤:
[0040]
s1:(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮的制备
[0041]
在2000ml反应瓶中投入1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮350g(1.16mol)和n,n,二甲基甲酰胺700g,将n,n-二甲基甲酰胺二甲缩醛152g(1.28mol)加入到反应瓶中,加热升温至125℃,保温反应2h。tlc检测:(石油醚60-90:乙酸乙酯=2:1)无1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮点,冷却降温至室温,将反应液加入到水中,搅拌3h后过滤,水洗,用3倍乙醇回流打浆后,冷却至室温,过滤,抽干,得(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮364.2g(1.02mol),摩尔收率:87.9%(以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮计)。
[0042]
s2:7-苄氧基-6-甲氧基-1h-喹啉-4-酮的制备
[0043]
在2000ml反应瓶中投入(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮360g(1.01mol)和乙醇720ml,搅拌加入二氧化硫脲218.4g(2.02mol)升温至80℃左右,保温搅拌5h。加入720g水中搅拌,降温至10-15℃,搅拌1h,过滤,水洗,鼓风干燥,得7-苄氧基-6-甲氧基-1h-喹啉-4-酮228g(0.81mol),摩尔收率:80.3%(以(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮计)。
[0044]
s3:4-氯-6-甲氧基-7-苄氧基喹啉的制备
[0045]
在反应瓶中投入7-苄氧基-6-甲氧基-1h-喹啉-4-酮220g(0.78mol)及二氯甲烷660ml,在35℃以下慢慢滴加三氯氧磷180g(0.78mol),滴加完毕后降温至25~30℃,开始滴加n,n-二异丙基乙胺101g(0.78mol),控制温度在40-45℃,加完后40-45℃保温1.5h。tlc检测:(石油醚60-90:乙酸乙酯=2:1)无7-苄氧基-6-甲氧基-1h-喹啉-4-酮,减压蒸馏除去大部分溶剂。降温至25℃以下,将反应液倒入冰水中,有大量棕黄色固体析出,抽滤,滤饼水洗至中性,70~80℃烘5h。在反应瓶中,投入4-氯-6-甲氧基-7-苄氧基喹啉粗品,加入二氯甲烷440g,搅拌升温至回流溶解,加入活性炭4g,搅拌30分钟,过滤,滤液减压蒸馏至剩约300g左右,降温至室温,析出固体,过滤,滤饼鼓风干燥,得高纯度4-氯-6-甲氧基-7-苄氧基喹啉183g(0.61mol),4-氯-6-甲氧基-7-苄氧基喹啉的高效液相色谱参见附图2,由液相色谱数据可知其纯度:99.79%,摩尔收率:78%(以7-苄氧基-6-甲氧基-1h-喹啉-4-酮计)。
[0046]
4-氯-6-甲氧基-7-苄氧基喹啉的核磁图谱参见附图3,其核磁数据为:1h nmr
(400mhz,j
6-dmso):δ8.61(s,ih),7.57-7.37(m,8h),5.32(s,2h),3.98(s,3h),与理论核磁数据一致;
[0047]
4-氯-6-甲氧基-7-苄氧基喹啉的碳谱参见附图4,其碳谱数据为:13
c nmr(100mhz,j
6-dmso):δ152.4,151.5,148.5,146.2,139.6,137.0,129.2,128.8,121.7,120.4,110.1,101.9,70.8,56.5,与理论碳谱数据一致。
[0048]
实施例2
[0049]
采用本发明方法制备4-氯-6-甲氧基-7-苄氧基喹啉,合成路线参见附图1,包括如下操作步骤:
[0050]
s1:(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮的制备
[0051]
在2000ml反应瓶中投入1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮300g(0.99mol),甲苯600g,n,n-二甲基甲酰胺二甲缩醛356g(0.98mol)加入到反应瓶中,加热升温至110℃,保温反应3h,tlc检测:(石油醚60-90:乙酸乙酯=2:1)无1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮点,冷却降温至室温,将反应液加入到水中,搅拌1h后过滤,水洗,用1倍甲苯回流打浆后,冷却至室温,过滤,抽干。得(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮315.7g(0.88mol)摩尔收率:88.5%(以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮计)。
[0052]
s2:7-苄氧基-6-甲氧基-1h-喹啉-4-酮的制备
[0053]
在2000ml反应瓶中投入(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮310g(0.87mol)、异丙醇930ml,搅拌加入连二亚硫酸钠196.8g(1.13mol),加入结束,升温至70℃保温搅拌5h;取样tlc检测:(石油醚60-90:乙酸乙酯=2:1)无(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮,加水930g,搅拌1h,过滤,水洗,鼓风干燥得7-苄氧基-6-甲氧基-1h-喹啉-4-酮203g(0.72mol)摩尔收率:83%(以(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮计)。
[0054]
s3:4-氯-6-甲氧基-7-苄氧基喹啉的制备
[0055]
在反应瓶中投入7-苄氧基-6-甲氧基-1h-喹啉-4-酮200g(0.71mol)和三氯氧磷1000g(6.52mol),在25~30℃,滴加二乙胺477g(6.52mol),控制温度在30~35℃,加完后40℃保温6h,tlc检测:(石油醚60-90:乙酸乙酯=2:1)无7-苄氧基-6-甲氧基-1h-喹啉-4-酮,减压蒸馏除去大部分三氯氧磷溶剂。
[0056]
降温至25℃以下,将反应液倒入冰水中,有大量棕黄色固体析出,抽滤,滤饼水洗至中性,过滤,滤饼鼓风干燥得高纯度4-氯-6-甲氧基-7-苄氧基喹啉172g(0.57mol),纯度99.73%,摩尔收率:80.7%(以7-苄氧基-6-甲氧基-1h-喹啉-4-酮计),4-氯-6-甲氧基-7-苄氧基喹啉的核磁、碳谱数据与实施例1中的一致。
[0057]
实施例3
[0058]
采用本发明方法制备4-氯-6-甲氧基-7-苄氧基喹啉,合成路线参见附图1,包括如下操作步骤:
[0059]
s1:(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮的制备
[0060]
在2000ml反应瓶中投入1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮300g(0.99mol/l),n,n-二甲基甲酰胺600g,n,n-二甲基甲酰胺二甲缩醛350g(2.96mol/l)加入到反应瓶中,加热升温至55℃,保温反应8h,tlc检测:(石油醚60-90:乙酸乙酯=2:1)无1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮点,冷却降温至5-10℃,将反应液加入到水中,搅拌3h后过滤,水洗,用3倍乙醇回流打浆后,冷却至室温,过滤,抽干。得(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮314g(0.88mol/l)摩尔收率:88.5%(以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮计)。
[0061]
s2:7-苄氧基-6-甲氧基-1h-喹啉-4-酮的制备
[0062]
在2000ml反应瓶中投入(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮310g(0.87mol/l)、甲醇:水(7:3混合溶液)620ml,搅拌分批加入硼氢化钠164g(4.33mol/l),加入结束,于20-30℃保温搅拌1.5h;取样tlc检测:(石油醚60-90:乙酸乙酯=2:1)无(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮,反应液加入到3100g水中,搅拌3h,过滤,水洗,鼓风干燥得7-苄氧基-6-甲氧基-1h-喹啉-4-酮217.8g,摩尔收率:89%(以(e)-1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)-3-(二甲基氨基)-2-丙烯-1-酮计)。
[0063]
s3:4-氯-6-甲氧基-7-苄氧基喹啉的制备
[0064]
在反应瓶中投入7-苄氧基-6-甲氧基-1h-喹啉-4-酮200g(0.71mol/l)和氯化亚砜253.7g(2.13mol/l),在30~35℃,滴加n,n-二异丙基乙胺275.6g(2.13mol/l),控制温度在15~25℃,加完后升温至90℃保温5h,tlc检测:(石油醚60-90:乙酸乙酯=2:1)无7-苄氧基-6-甲氧基-1h-喹啉-4-酮,减压蒸馏除去大部分氯化亚砜溶剂。
[0065]
降温至25℃以下,加二氯甲烷400g搅拌,将反应液倒入冰水中,搅拌分层,二氯甲烷层水洗,有机层加活性炭4g搅拌1小时,过滤,滤液减压蒸馏至剩约300g,降温至5-10℃析晶有大量棕黄色固体析出,过滤,滤饼鼓风干燥得高纯度4-氯-6-甲氧基-7-苄氧基喹啉183.5g,纯度99.97%,摩尔收率:86.1%(以7-苄氧基-6-甲氧基-1h-喹啉-4-酮计),4-氯-6-甲氧基-7-苄氧基喹啉的核磁、碳谱数据与实施例1中的一致。
[0066]
本发明公开了一种4-氯-6-甲氧基-7-苄氧基喹啉的制备方法,以1-(4-(苄氧基)-5-甲氧基-2-硝基苯基)乙酮为起始原料,经过烯胺化、还原环合、氯代反应制备得4-氯-6-甲氧基-7-苄氧基喹啉,反应原料易得,不需高温高压,常压即可操作,反应条件温和,工艺简洁,操作简便,收率高,成本低,适合工业化放大生产;制备7-苄氧基-6-甲氧基-1h-喹啉-4-酮采用强还原环合剂二步一锅法:由原硝基还原为氨基后再经过环合试剂进行环合得到目标7-苄氧基-6-甲氧基-1h-喹啉-4-酮,不需经高温高压和通入氢气反应等高危工艺操作,即可制备得到7-苄氧基-6-甲氧基-1h-喹啉-4-酮,显著提高操作安全性。
[0067]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1