具有手性可控的偶氮苯聚合物超分子组装体及其手性调控方法
1.本发明属于高分子合成技术领域,涉及一种具有手性可控的偶氮苯聚合物超分子组装体及其手性调控方法,具体通过调整手性中心和偶氮苯之间距离来进行超分子手性调控。
背景技术:
2.手性是自然界的基本属性,在自然界中普遍存在,在超分子领域,一些组装模块在多种非共价相互作用下进行手性排列,形成具有螺旋结构的组装体,使原本不具有手性的分子或基团产生手性信号、或者使原有手性以非线性的形式被放大,这种现象称为超分子手性。人们可以通过改变分子间的非共价相互作用来调控超分子手性,例如可以通过改变溶剂、温度、离子以及ph等条件来达到调控超分子手性的目的。除此之外,对手性组装基元结构的改变同样可以达到手性调控的目的。尽管此前在小分子液晶体系中就有通过调整手性中心和液晶基元距离的奇偶变化来达到调控手性的报道,但目前还没有在聚合物体系中调整手性中心和组装基元距离奇偶变化进行聚合物组装体手性调控的报道。
技术实现要素:
3.本发明旨在利用调整手性中心和组装基元距离奇偶变化的策略调控超分子手性,提供一种具有手性可控的偶氮苯聚合物超分子组装体及其手性调控方法。
4.按照本发明的技术方案,所述具有手性可控的偶氮苯聚合物超分子组装体,由手性侧链型偶氮苯聚合物在良溶剂-不良溶剂的混合体系中进行超分子手性组装得到;所述手性侧链型偶氮苯聚合物为pazo-l-m或pazo-d-m,由手性偶氮苯单体经raft聚合(可逆加成-断裂链转移聚合)得到,其中,m是指手性中心与偶氮苯之间距离,为3或6-16中任一整数;所述手性偶氮苯单体的结构式如下述任一所示:
[0005][0006]
其中,x为2-12中任一整数。
[0007]
本发明设计利用raft聚合获得了手性中心到偶氮苯不同距离的手性侧链型聚合物,这个距离是奇偶交替变化的,接下来通过后组装策略,聚合物在良溶剂-不良溶剂的混合体系中进行超分子手性组装。手性中心到偶氮苯的距离以及良溶剂-不良溶剂比例都可以用来调控该偶氮苯聚合物组装体的超分子手性和聚合物在溶解状态下的大分子手性。
[0008]
优选的,m为3、6、7、8、9或10;x=2、3、4、5或6。
[0009]
进一步的,所述良溶剂-不良溶剂的混合体系中,良溶剂为1,2-二氯乙烷或四氢呋喃,不良溶剂为甲基环己烷或乙醇;具体的,所述良溶剂-不良溶剂的混合体系为1,2-二氯乙烷(dce)-甲基环己烷(mch)混合体系或四氢呋喃(thf)-乙醇(etoh)混合体系,优选为dce-mch。
[0010]
进一步的,每3ml良溶剂-不良溶剂的混合体系中,手性侧链型偶氮苯聚合物的质量为0.05-0.2mg。
[0011]
进一步的,改变混合体系中良溶剂和不良溶剂的比例,可以调控组装体的形貌和手性表达。
[0012]
进一步的,所述手性侧链型偶氮苯聚合物中的重复单元为25-40个,最优选为32个。
[0013]
进一步的,所述raft聚合在引发剂存在下、有机溶剂中进行,手性偶氮苯单体与raft试剂的摩尔比为20-80﹕1,优选为50﹕1。
[0014]
进一步的,所述引发剂为偶氮二异丁腈、偶氮二异庚腈或偶氮二异丁酸二甲酯,优选偶氮二异丁腈(aibn);所述有机溶剂为四氢呋喃或苯甲醚,优选四氢呋喃。
[0015]
进一步的,所述raft试剂为cpdn(二硫代萘甲酸异丁腈酯)。
[0016]
进一步的,所述raft聚合的温度为60-80℃,时间为3-6h;优选70℃反应4小时。
[0017]
进一步的,所述手性偶氮苯单体的制备方法如下:
[0018]
s1:以对甲氧基苯胺、苯酚为原料制备化合物1,化合物1与卤素醇反应得到化合物2;
[0019]
s2:对手性乳酸甲酯进行羟基保护后,水解得到化合物4;
[0020]
s3:化合物1与化合物4,或化合物2和化合物4通过酯化反应得到化合物5;
[0021]
s4:化合物5脱去羟基保护基,得到化合物6;
[0022]
s5:化合物6与甲基丙烯酰氯在保护气氛下反应,得到所述手性偶氮苯单体(化合物7);
[0023]
其中,所述步骤s1和s2的顺序不限。
[0024]
具体的,所述步骤s3中,化合物1与化合物4酯化反应,得到mazo-l-3或mazo-d-3;化合物2与化合物4酯化反应,得到mazo-l-m或mazo-d-m。
[0025]
进一步的,所述卤素醇为碳原子个数为3-12的卤素醇,可以选用3-溴丙醇、4-溴-1-丁醇、5-溴-1-戊醇、6-溴-1-己醇等。
[0026]
进一步的,所述步骤s2中,采用叔丁基二苯基氯硅烷(tbdpscl)进行羟基保护,反应得到化合物3;化合物3在氢氧化锂的四氢呋喃和水的混合溶液水解(脱甲酯)得到化合物4。
[0027]
进一步的,所述步骤s4中,采用四丁基氟化铵(tbaf)和醋酸混合溶液脱去羟基保护基。
[0028]
进一步的,所述保护气氛为氩气、氦气、氮气,优选氩气。
[0029]
具体的,本发明具有手性可控的偶氮苯聚合物超分子组装体的制备方法可以如下:
[0030]
1、合成手性偶氮苯单体
[0031]
将原料对甲氧基苯胺、去离子水、浓盐酸倒入烧杯中,在冰水浴的条件搅拌,随后极其缓慢滴加亚硝酸钠水溶液,这样就成功的制得重氮盐水溶液,溶液体系呈黑红色;制备重氮盐水溶液的同时,将苯酚、氢氧化钠、碳酸氢钠溶于去离子水中,在冰水浴条件下将上述制的重氮盐水溶液缓慢滴加到苯酚溶液中,反应时间4-6h。反应结束之后,抽滤,冲洗滤饼,随后烘干,乙醇重结晶,得到化合物1;
[0032]
将上述化合物1加入到反应瓶中,然后加入碳酸钾、n,n二甲基甲酰胺,于70-90℃条件搅拌,随后加入碘化钾催化剂,缓慢滴加2-溴乙醇,反应过夜;次日冷却至室温,抽滤,倒入水中,乙酸乙酯萃取,水洗乙酸乙酯相,干燥,抽滤,旋蒸乙醇进行重结晶,得到化合物2;
[0033]
l-乳酸甲酯、咪唑溶于二氯甲烷中,随后在0℃条件滴加叔丁基二苯基氯硅烷,然后移至室温反应过夜;反应结束,盐酸酸化,然后用饱和碳酸氢钠,饱和食盐水各洗涤一遍有机相;干燥,抽滤,旋蒸,柱层析,得到化合物3;
[0034]
化合物3溶于四氢呋喃,在冰水浴条件下加入冰的氢氧化锂,搅拌,tlc跟踪反应进程,反应结束用盐酸调ph约等于3,乙酸乙酯萃取,食盐水洗涤,干燥,抽滤,旋蒸,得到化合物4;
[0035]
化合物4溶于的二氯甲烷,然后加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)和4-二甲氨基吡啶(dmap),随后将化合物4加入到上述溶液。次日反应结束后加入去离子水淬灭反应,水和食盐水洗涤有机相,干燥,抽滤,旋蒸,柱层析,得到化合物5;
[0036]
将化合物5溶于四氢呋喃中,随后将四丁基氟化铵和醋酸的混合溶液滴加到四氢呋喃溶液中,tlc监测反应结束,加水淬灭,乙酸乙酯萃取,食盐水洗涤有机相,干燥,抽滤,旋蒸,柱层析,得到化合物6;
[0037]
将三乙胺、化合物6加入到四氢呋喃溶液中。在氩气下回流下滴加甲基丙烯酰氯,10h后反应结束,抽滤除去固体不溶物,旋蒸除去四氢呋喃,乙酸乙酯溶解,饱和碳酸氢钠,水,食盐水洗涤有机相,干燥,抽滤,旋蒸,柱层析,得到化合物7。
[0038]
2、单体的聚合
[0039]
将单体(比如mazo-l-6)、raft试剂(比如cpdn)、引发剂(比如aibn)和溶剂(比如thf)放入到安瓿瓶中,安瓿瓶在经过除氧后,封管。在控制聚合温度60-80℃聚合3-6h,聚合淬灭聚合,然后破管,在甲醇中沉降两次得到聚合物pazo-l-6。
[0040]
3、聚合物组装体
[0041]
首先聚合物先溶解在良溶剂中(比如1,2-二氯乙烷),然后将聚合物溶液滴加到不良溶剂(比如甲基环己烷)中,随后摇晃混合溶液,即可得到聚合物组装体。
[0042]
进一步的,沉降溶剂除甲醇外还可选乙醇、石油醚,优选甲醇。
[0043]
通常来说,每一步反应完成之后都可以进行纯化,目的是得到纯度更高的产物,所述纯化步骤包括(但不限于)色谱法、重结晶法、溶解/沉淀分离法、过滤法等。
[0044]
本发明的第二方面提供了上述偶氮苯聚合物超分子组装体的手性调控方法,采用
以下方式中的一种或多种:
[0045]
调整手性中心与偶氮苯之间距离m的奇偶变化,
[0046]
调整良溶剂-不良溶剂的混合体系中良溶剂与不良溶剂的比例,
[0047]
超分子手性组装过程中加热。
[0048]
本发明的第三方面提供了上述偶氮苯聚合物超分子组装体在手性模板、手性识别、圆偏振发光或不对称催化等领域中的应用。
[0049]
本发明的技术方案相比现有技术具有以下优点:
[0050]
本发明合成了具有手性可控的偶氮苯聚合物超分子组装体,首次尝试在聚合物体系中利用调控手性中心到偶氮苯距离的奇偶变化,来调控聚合物在混合溶剂中组装体的超分子手性;
[0051]
本发明利用手性中心到偶氮苯距离的奇偶变化,可以获得螺旋方向相反的组装体,也可以调控组装体的光学活性,可以用作避免对映体的合成新手段。
附图说明
[0052]
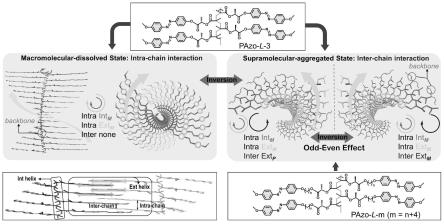
图1为手性中心到偶氮苯不同距离聚合物在溶解状态下和组装状态下的侧链偶氮苯的螺旋方向示意图。
[0053]
图2为手性偶氮苯单体合成路线。
[0054]
图3为手性中心到偶氮苯不同距离的手性单体的核磁图。
[0055]
图4为手性中心到偶氮苯不同距离的手性单体的手性hplc图。
[0056]
图5为聚合物的gpc流出曲线。
[0057]
图6为聚合物pazo-l-3在溶解状态下和组装状态下的圆二色谱图和紫外谱图。(a)为在良溶剂thf中;(b)为在混合溶剂(dce/mch)中;(c)为从溶解到组装再到热处理;(d)为手性多重反转开关。
[0058]
图7为热处理前后组装体随时间变化的圆二色谱图和紫外谱图;(a)热处理前;(b)热处理后。
[0059]
图8为聚合物pazo-l-3在良溶剂thf中进行循环使用365nm和435nm光照射的圆二色谱图和紫外谱图。
[0060]
图9为手性中心到偶氮苯距离逐渐增加的聚合物在良溶剂中圆二色谱图和紫外谱图以及vcd谱图。
[0061]
图10为手性中心到偶氮苯距离逐渐增加的l构型聚合物在混合溶剂(dce/mch)中的圆二色谱图和紫外谱图。
[0062]
图11为手性中心到偶氮苯距离逐渐增加的l构型和d构型聚合物在混合溶剂(dce/mch)中的圆二色谱图和紫外谱图。
[0063]
图12为手性中心到偶氮苯不同距离的聚合物在混合溶剂(dce/mch)中形成的组装体的tem图。
具体实施方式
[0064]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0065]
化学试剂:
[0066]
l-乳酸甲酯,98%,adamas;
[0067]
d-乳酸甲酯,98%,adamas;
[0068]
四丁基氟化铵,1.0m in thf,aladdin;
[0069]
叔丁基二苯基氯硅烷,98%,amethyst;
[0070]
2-溴乙醇,98%,aladdin;
[0071]
3-溴-1-丙醇,93%,aladdin;
[0072]
4-溴-1-丁醇,80%,aladdin;
[0073]
5-溴-1-戊醇,90%,aladdin;
[0074]
6-溴-1-己醇,95%,acros;
[0075]
四丁基氟化铵,1.0m in thf,aladdin;
[0076]
1,2-二氯乙烷,99%,greagent;
[0077]
甲基环己烷,99%,adamas;
[0078]
1,2-二氯乙烷,99%,greagent;
[0079]
苯酚,ar,aladdin;
[0080]
甲基丙烯酸,99%,aladdin;
[0081]
cpnd,定制合成;
[0082]
偶氮二异丁腈,使用前用乙醇重结晶两次;
[0083]
四氢呋喃,99.5%,南京化学试剂有限公司;
[0084]
乙醇,分析纯,江苏强盛功能化学股份有限公司;
[0085]
甲基丙烯酰氯,95%,aladdin;
[0086]
盐酸,分析纯,江苏强盛功能化学股份有限公司;
[0087]
亚硝酸钠,分析纯,江苏强盛功能化学股份有限公司;
[0088]
1,4-二氧六环,分析纯,江苏强盛功能化学股份有限公司;
[0089]
碘化钾,分析纯,江苏强盛功能化学股份有限公司;
[0090]
三乙胺,分析纯,江苏强盛功能化学股份有限公司;
[0091]
无水硫酸钠,98%,国药集团化学试剂有限公司;
[0092]
碳酸钾;分析纯,江苏强盛功能化学股份有限公司;
[0093]
氢氧化钠;分析纯,江苏强盛功能化学股份有限公司;
[0094]
碳酸氢钠;分析纯,江苏强盛功能化学股份有限公司;
[0095]
乙酸乙酯,99.5%,江苏强盛功能化学股份有限公司;
[0096]
石油醚,分析纯,江苏强盛功能化学股份有限公司;
[0097]
乙醚,分析纯,江苏强盛功能化学股份有限公司;
[0098]
测试仪器及条件:
[0099]
凝胶渗透色谱(gpc):分子量和分子量分布使用带有tosoh tskgel superhm-m的凝胶渗透色谱仪,属于自动进样式,聚苯乙烯作为标样计算聚合物分子量,四氢呋喃作为流动相,流速为0.65ml/min,温度为40℃。
[0100]
核磁共振氢谱(1h-nmr):使用bruker 300mhz核磁仪,以cdcl3为溶剂,tms为内标,室温下测定。
[0101]
透射电子显微镜(tem):使用hitachi ht 7700透射电子显微镜,加速电压为120kv。
[0102]
圆二色性(cd):使用日本jasco j-815圆二色光谱仪,20℃测量,扫描速度为200nm/min,扫描范围为250~600nm,带宽为2nm。
[0103]
本发明先合成手性中心到偶氮苯不同距离的手性单体,随后利用raft聚合获得了手性侧链型偶氮苯聚合物,接下来通过后组装策略,聚合物在良溶剂-不良溶剂的混合体系中进行超分子手性组装。手性中心到偶氮苯的距离以及良溶剂-不良溶剂比例都可以用来调控该偶氮苯聚合物组装体的超分子手性和聚合物在溶解状态下的大分子手性。图1为本发明示意图,从图中可以看出手性中心到偶氮苯的距离m对组装体有着决定性影响。
[0104]
实施例1手性偶氮苯单体的合成
[0105]
参见图2,为手性偶氮苯单体的合成路线图。
[0106]
以单体mazo-l-6的合成为例。将原料对甲氧基苯胺(12.1g)、去离子水(80ml可加入少量乙醇,促进溶解),和30ml的浓盐酸倒入烧杯中,在冰水浴的条件下控制温度为0-3℃,搅拌30min。随后极其缓慢滴加30ml的nano2(7g)水溶液,滴加完毕后继续反应30min,这样就成功的制的重氮盐水溶液,溶液体系呈黑红色。制备重氮盐水溶液的同时,将苯酚(16g),氢氧化钠(8g),碳酸氢钠(8.4g)溶于350ml的去离子水中,在冰水浴条件下将上述制的重氮盐水溶液缓慢滴加到上述的苯酚混合物溶液中,重氮盐的滴加过程中,反应液逐渐从无色变成浅黄色,随着时间的进行,黄色逐渐加深,并且有些偏红,反应时间4.5h。反应结束之后,抽滤得到不溶的偶氮苯固体,滤饼用大量的去离子水冲洗,随后烘干,选用乙醇进行重结晶即可得到纯净的化合物1;
[0107]
将上述化合物1(8g)加入到反应瓶中,然后加入碳酸钾(36.7g),n,n二甲基甲酰胺(100ml),于80℃油浴锅中搅拌30min。随后加入ki催化剂,缓慢滴加2-溴乙醇,反应过夜。次日反应结束,冷却至室温,抽滤除去大量碳酸钾,然后倒入水中,用乙酸乙酯萃取三次,然后用水洗乙酸乙酯相三次除去n,n二甲基甲酰胺,随后无水硫酸钠干燥有机相,抽滤,旋蒸,固体产物用乙醇进行重结晶,即可得到化合物2;
[0108]
l-乳酸甲酯(1.9g),咪唑(3.26g)共同溶于60ml的二氯甲烷中,随后在0℃条件下缓慢滴加叔丁基二苯基氯硅烷(tbdpscl,6.14ml),然后移至室温反应过夜。反应结束,加1m盐酸酸化,然后用饱和碳酸氢钠,饱和食盐水各洗涤一遍有机相。无水硫酸钠干燥,抽滤,旋蒸,选用石油醚(pe):乙酸乙酯(ea)=30:1进行柱层析,即可得到目标产物化合物3;
[0109]
取化合物3(6g)溶于270ml的四氢呋喃(thf)中,在冰水浴条件下加入冰的氢氧化锂(0.2m,175ml),搅拌,tlc跟踪反应进程,反应6h,随后用1m盐酸调ph约等于3,然后用ea萃取二次,食盐水洗涤有机相一次,无水硫酸钠干燥,抽滤,旋蒸即可得到目标产物化合物4;
[0110]
化合物2(6g)溶于100ml的二氯甲烷(dcm),然后加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci,7.1g)和4-二甲氨基吡啶(dmap,0.49g),随后在0℃的条件下缓慢滴加化合物4(3.5g)的二氯甲烷溶液,反应过夜,次日反应结束后加入去离子水淬灭反应,然后用水和食盐水洗涤有机相,无水硫酸钠干燥,抽滤,旋蒸,选用pe:ea=15:1进行柱层析,即可得到目标产物化合物5;
[0111]
将化合物5(3.4g)溶于70ml thf中,随后将四丁基氟化铵(tbaf,1mol/ml,7ml)和醋酸(400微升)的混合溶液滴加到thf溶液中,tlc监测30min后反应结束,然后加水淬灭,选
用ea萃取,食盐水洗涤有机相,无水硫酸钠干燥,抽滤,旋蒸,选用pe:ea=3:1进行柱层析,即可得到目标产物化合物6;
[0112]
将三乙胺(4ml),化合物6(2.2g)加入到thf(70ml)溶液中。在氩气下回流将甲基丙烯酰氯(3ml)缓慢滴加到上述thf溶液中,10h后反应结束,抽滤除去固体不溶物,旋蒸除去thf,然后选用ea溶解,随后用饱和碳酸氢钠,水,食盐水洗涤,无水硫酸钠干燥抽滤,旋蒸,选用pe:ea=15:1进行柱层析,即可得到目标产物化合物7。
[0113]
当卤素醇选用3-溴丙醇、4-溴-1-丁醇、5-溴-1-戊醇、6-溴-1-己醇或者不加卤素醇时,可以得到其它手性中心到偶氮苯不同距离的单体。
[0114]
不同单体的核磁表征见图3,手性hplc表征见图4。
[0115]
实施例2单体的聚合(以聚合物pazo-l-3为例):
[0116]
将mazo-l-3(220.8mg,0.60mol),cpdn(3.26mg,0.012mmol),aibn(0.656mg,0.004mmol)和无水thf(1.0ml)放入到一个5ml的安瓿瓶中,安瓿瓶在经过三次冻融循环除氧后,在氩气气氛下进行火焰密封。在70℃聚合4h,聚合结束冰水浴降温淬灭聚合,然后破管,在甲醇中沉降两次得到聚合物pazo-l-3。其中单体:cpdn:aibn的摩尔比为150:3:1。
[0117]
所得聚合物的gpc表征见图5。
[0118]
实施例3聚合物组装体的制备的一般过程
[0119]
将聚合物首先配制成1mg/ml的1,2-二氯乙烷(dce)溶液,若制备体积比为dce/mch=0.2/2.8的组装体溶液,具体步骤为,取0.1ml的配制好的聚集体溶液加入到比色皿中,随后补加0.1ml的dce纯净溶剂,保证dce共0.2ml,然后将2.8ml的甲基环己烷(mch)缓慢滴加到上述比色皿中,随着摇晃比色皿使之形成均匀的组装体溶液。其它体积比的组装体溶液制备过程类似,但保证每3ml混合溶液中共含有0.1mg的偶氮苯聚合物。
[0120]
实施例4手性控制的表征
[0121]
手性中心到偶氮苯的距离对聚合物的手性表达起着控制作用。图6为聚合物pazo-l-3的表征,其在溶解状态下表现出负的cotton效应,一旦加入不良溶剂,使其形成组装体,则变成了正的cotton效应,也就是说从溶解态到组装态发生了一次的手性反转现象。在dce/mch=0.6/2.4时,在对组装体溶液进行热处理之后,其发生了第二次手性反转现象,也就是说聚合物pazo-l-3从溶解态—组装态—热处过程共经历了两次手性反转现象,利用这个多次手性反转的实验现象,成功构建了一个一次循环两次反转的手性反转开关(图6d)。对于聚合物pazo-l-3,通过对外界实验条件的控制,实现了对其溶解态大分子手性以及组装态超分子手性的控制。
[0122]
对于聚合物pazo-l-3,在dce/mch=0.6/2.4时其表现出正的cotton效应,当对其进行热处理之后,则变成了负的cotton效应,发生了手性反转现象。这两种状态时均是组装态,测试这两种状态下组装体随时间变化的圆二色谱,如图7所示,随着时间的增加,未热处理的组装体的cd信号逐渐降低直至消失,而经过热处理的组装体的cd信号随着时间的增加几乎不变,所以前一种状态可以认为是动力学控制的产物,而经过热处理之后则变成了热力学控制的产物。所以组装状态下的手性反转可以认为是动力学控制产物到热力学控制产物的手性反转。
[0123]
偶氮苯具有光致异构特性,可以利用其顺反异构导致的共面结构和非共面结构变化,来构建手性有无的开关,利用此性质,如图8所示,聚合物pazo-l-3在溶解状态下,循环
的使用365nm和435nm的光照射,使之发生光致异构,成功的构建了一个至少可以循环五次手性开关。
[0124]
随后对手性中心到偶氮苯距离逐渐增加的聚合物进行了手性表征。如图9所示,对于手性中心到偶氮苯距离为6,7,8,9,10的聚合物其在溶解状态下在偶氮苯吸收区域均无cd信号,而通过振动圆二色谱(vcd)表征可知主链此时依然存在着螺旋构象,可以解释为主链和偶氮苯之间存在着较长的柔性链,即使主链是螺旋构象也不能够带动侧链偶氮苯的螺旋排列。随后对聚合物组装体进行了圆二色谱(cd)测试,如图10所示,可以看到pazo-l-6在组装状态下表现出负的cotton效应;pazo-l-7在组装状态下表现出正的cotton效应;pazo-l-8在组装状态下表现出负的cotton效应;pazo-l-9在组装状态下表现出正的cotton效应;pazo-l-10在组装状态下表现出负的cotton效应;从测试结果来看,随着距离的奇偶变化,组装体的螺旋方向发生了明显的奇偶交替反转,而且手性信号的强度也存在着奇偶的变化,偶数距离的聚合物组装体具有更强的手性信号表达。d构型的聚合物组装体相较于l构型的聚合物组装体具有镜像的cd信号(图11)。通过对聚合物组装体的形貌表征,如图12所示,可以看出随着不良溶剂比例的逐渐增加,所有聚合物组装体的形貌都是从球形形貌逐渐变成无规聚集体。本发明偶氮苯聚合物超分子组装体可以利用调整手性中心到偶氮苯的距离实现了手性的高效调控,这个距离可以对手性螺旋方向和手性信号大小进行调控;此外外界条件例如混合溶剂比例、热处理等也可以用做手性调控的手段。
[0125]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1