一种乙烯基异噁唑烷衍生物的制备方法
1.本发明涉及有机化学合成技术领域,具体为一种乙烯基异噁唑烷衍生物的制备方法。
背景技术:
2.异噁唑环类化合物作为核苷、碳水化合物、pna、氨基酸和类固醇类似物的模拟物,以及其抗癌,抗肿瘤,抗菌,抑制细胞周期分裂蛋白25(cdc25a/cdc25b)酶和含sh2结构域蛋白酪氨酸磷酸酶1(shp-1)等广泛的生物活性而倍受关注。研究表明,将几个不同官能团或者分子骨架引入到某些分子中就行修饰,可改善母体分子生物活性,以便寻找高活性医药前体化合物。近年来,人们对异噁唑烷和含有异噁唑烷环的化合物的研究越来越多,涉及异噁唑烷环的新型化合物及其反应已被用于实现全合成或获得具有生物活性的化合物。
3.本发明以异噁唑烷为分子骨架,引入乙烯基得到一种新型的2-乙烯基异噁唑烷衍生物,并通过铜催化的方法合成得到多种类型2-乙烯基异噁唑烷衍生物,丰富异噁唑环类化合物库。
技术实现要素:
4.本发明的目的在于提供一种乙烯基异噁唑烷衍生物的制备方法,制备方法简单,且可以通过优化反应条件达到反应条件温和,产率高、纯度高的效果。
5.本发明的目的可以通过以下技术方案实现:
6.一种乙烯基异噁唑烷衍生物的制备方法,包括如下步骤:有机溶剂在添加有铜催化剂和碱时,将o-丙炔基肟醚化合物与磺酰基叠氮搅拌反应,得到2-乙烯基异噁唑烷衍生物;o-丙炔基肟醚化合物的结构式为磺酰基叠氮的结构式为;2-乙烯基异噁唑烷衍生物的结构式为
7.其中,o-丙炔基肟醚化合物、磺酰基叠氮和2-乙烯基异噁唑烷衍生物结构式中的r1、r2、r3独立地选自苯基、取代苯基、萘基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、卤素、卤代c
1-c6烷基或卤代c
1-c6烷氧基中的任意一种;
8.所述铜催化剂为醋酸铜、氯化铜、溴化铜、乙酰丙酮铜、三氟乙酸铜、三氟甲磺酸铜、氧化铜、碘化亚铜、溴化亚铜、氯化亚铜、噻吩-2-甲酸铜、醋酸亚铜中的任意一种;
9.所述碱为三乙胺、正三丁胺、三叔丁胺、2-氟吡啶、2-氯吡啶、二异丙基乙胺、吡啶、2-甲基吡啶、氢氧化钠中的任意一种;
10.所述有机溶剂为甲醇、乙醇、乙腈、四氢呋喃、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、氯苯、苯、二甲苯中的一种。
11.作为本发明进一步的方案:所述o-丙炔基肟醚化合物、铜催化剂、碱的摩尔比为1:0.05-0.4:0.1-2;所述o-丙炔基肟醚化合物、磺酰基叠氮摩尔比为1:1-3。
12.作为本发明进一步的方案:搅拌反应的温度为0-60℃且反应时间为0.5-8小时。
13.本发明的有益效果:
14.本发明使用铜催化剂、使用胺类化合物作为碱,可由o-丙炔基肟醚化合物、磺酰基叠氮反应,一步得到2-乙烯基异噁唑烷衍生物,具有反应条件温和,产率高、纯度高、原子经济性高等诸多优点,为2-乙烯基异噁唑烷衍生物提供了全新路线和新思路,可在合成中间体、药物中间体、农药中间体等领域中发挥重要作用,在工业和科研上具有良好的应用价值和潜力。
附图说明
15.为了便于本领域技术人员理解,下面结合附图对本发明作进一步的说明。
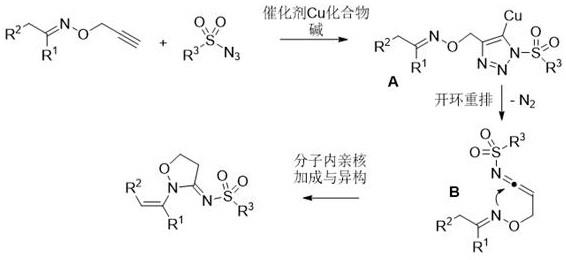
16.图1为本发明o-丙炔基肟醚化合物与的磺酰基叠氮的反应机理图。
具体实施方式
17.下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
18.如图1所示,一种乙烯基异噁唑烷衍生物的制备方法,包括如下步骤:有机溶剂在添加有铜催化剂和碱时,将o-丙炔基肟醚化合物与磺酰基叠氮搅拌反应,得到2-乙烯基异噁唑烷衍生物;o-丙炔基肟醚化合物的结构式为磺酰基叠氮的结构式为;2-乙烯基异噁唑烷衍生物的结构式为
19.其中,o-丙炔基肟醚化合物、磺酰基叠氮和2-乙烯基异噁唑烷衍生物结构式中的r1、r2、r3独立地选自苯基、取代苯基、萘基、c
1-c6烷基、c
2-c6烯基、c
1-c6烷氧基、卤素、卤代c
1-c6烷基或卤代c
1-c6烷氧基中的任意一种;
20.铜催化剂为醋酸铜、氯化铜、溴化铜、乙酰丙酮铜、三氟乙酸铜、三氟甲磺酸铜、氧化铜、碘化亚铜、溴化亚铜、氯化亚铜、噻吩-2-甲酸铜、醋酸亚铜中的任意一种;
21.碱为三乙胺、正三丁胺、三叔丁胺、2-氟吡啶、2-氯吡啶、二异丙基乙胺、吡啶、2-甲基吡啶、氢氧化钠中的任意一种;最优选为三乙胺;
22.有机溶剂为甲醇、乙醇、乙腈、四氢呋喃、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、氯苯、苯、二甲苯中的一种;优选为乙腈;
23.o-丙炔基肟醚化合物、铜催化剂、碱的摩尔比为1:0.05-0.4:0.1-2;o-丙炔基肟醚化合物、磺酰基叠氮摩尔比为1:1-3;
24.搅拌反应的温度为0-60℃且反应时间为0.5-8小时;
25.c
1-c6烷基指具有1-6个碳原子的直链或支链烷基,其包括了c1烷基、c2烷基、c3烷基、c4烷基、c5烷基或c6烷基,可为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基或正己基等;
26.c
1-c6烷氧基是指c
1-c6烷基与o原子相连后的基团;
27.卤素的含义是指卤族元素,可为f、cl、br或i;
28.卤代c
1-c6烷基的含义是指被卤素取代的c
1-c6烷基,可为三氟甲基、五氟乙基、二氟甲基、氯甲基等;
29.卤代c
1-c6烷氧基的含义是指被卤素取代的c
1-c6烷氧基,可为三氟甲氧基、五氟乙氧基、二氟甲氧基、氯甲氧基等;
30.制备步骤具体是:将o-丙炔基肟醚化合物与磺酰基叠氮混合进行反应,在反应过程中,首先磺酰基叠氮与端炔化合物发生1,3-偶极环加成反应得到三氮唑复合物,接着发生开环重排,再与o-丙炔基肟醚化合物发生分子内的亲核加成、异构化得到2-乙烯基异噁唑烷衍生物;
31.反应在铜催化剂的催化和碱的作用下进行,在铜催化剂和碱作用下,o-丙炔基肟醚化合物与的磺酰基叠氮的反应机理如图1;
32.首先在铜催化剂、碱的作用下o-丙炔基肟醚化合物与磺酰基叠氮发生1,3-偶极环加成得到铜的复合物三氮唑a,接着发生开环重排,得到烯酮亚胺中间体b,烯酮亚胺中间体b发生分子内的亲核进攻,同时碳氮双键发生异构,最后得到2-乙烯基异噁唑烷衍生物;
33.铜催化剂包括醋酸铜、氯化铜、溴化铜、乙酰丙酮铜、三氟乙酸铜、三氟甲磺酸铜、氧化铜、碘化亚铜、溴化亚铜、氯化亚铜、噻吩-2-甲酸铜、醋酸亚铜中的任意一种或多种,优选碘化亚铜或氯化亚铜,最优选为碘化亚铜;
34.o-丙炔基肟醚化合物与铜催化剂的摩尔比为1:0.05-0.4;
35.丙炔基肟醚化合物与碱的摩尔比为1:0.1-2;
36.o-丙炔基肟醚化合物与有机溶剂的比例为1mmol:5-15ml,例如1mmol:5ml、1mmol:8ml、1mmol:10ml、1mmol:12ml或1mmol:15ml;
37.反应结束后还包括后处理步骤,后处理包括萃取、浓缩、结晶、重结晶、柱层色谱提纯中的任何一种处理手段或多种处理手段的组合;
38.作为一种例举性的后处理手段,例如可为:将反应体系冷却至室温,旋转蒸发仪蒸除溶剂,残留物过200-300目硅胶柱,以乙酸乙酯/石油醚为洗脱剂,其中乙酸乙酯与石油醚的体积比为1:5-15,从而得到2-乙烯基异噁唑烷衍生物目标产物;
39.作为另一种例举性的后处理手段,例如可为:反应完全后,将反应体系自然冷却至室温,加入等体积比的乙酸乙酯和饱和食盐水的混合液,振荡萃取2-4次,收集有机层、干燥,旋转蒸发浓缩,得粗产物,将粗产物过200-300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1:5-10,从而得到2-乙烯基异噁唑烷衍生物目标产物。
40.实施例1
41.向乙腈中加入o-丙炔基肟醚化合物和磺酰基叠氮、碘化亚铜、三乙胺,然后室温下搅拌密封反应4小时。
42.其中,o-丙炔基肟醚化合物与碘化亚铜的摩尔比为1:0.10;o-丙炔基肟醚化合物
与三乙胺的摩尔比为1∶1.0;0-丙炔基肟醚化合物化合物与磺酰基叠氮的摩尔比为1∶1.5;以及以毫摩尔计的o-丙炔基肟醚化合物化合物与以毫升计的乙腈的比为1∶5;
43.反应结束后,将反应体系自然冷却至室温,旋转蒸发浓缩,得粗产物,将粗产物过300-400目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1∶5,从而得到为浅黄色粘稠液体的目标产物2-乙烯基异噁唑烷衍生物,产率为92%,纯度为99.0%;
44.核磁共振:1hnmr(400mhz,氘代氯仿cdcl3)δ7.76(d,j=7.8hz,1h),7.64-7.60(m,1h),7.56-7.54(m,2h),7.44-7.40(m,2h),7.27-7.17(m,5h),7.15-7.11(m,1h),7.02-7.03(m,1h),6.63(d,j=8.6hz,1h),4.52(s,2h)。
45.13
cnmr(400mhz,氘代氯仿cdcl3)δ7.59(d,j=8.4hz,2h),7.31(s,5h),7.18(d,j=8.0hz,2h),5.61(s,1h),5.51(s,1h),4.40(t,j=8.2hz,2h),3.71(t,j=8.2hz,2h),2.38(s,3h);
46.13
c nmr(100mhz,cdcl3)δ160.4,142.6,140.7,139.6,134.1,129.3(2c),129.2,128.5(2c),126.6(2c),126.4(2c),112.4,67.4,34.6,21.6。
47.实施例2
48.向乙腈中加入o-丙炔基肟醚化合物和磺酰基叠氮、碘化亚铜、三乙胺,然后升温至30℃,并在该温度下搅拌密封反应6小时;
49.其中,o-丙炔基肟醚化合物与碘化亚铜的摩尔比为1∶0.15;o-丙炔基肟醚化合物与三乙胺的摩尔比为1∶1.2;o-丙炔基肟醚化合物化合物与磺酰基叠氮的摩尔比为1∶1.2;以及以毫摩尔计的o-丙炔基肟醚化合物与以毫升计乙腈的比为1∶7;
50.反应完全后,将反应体系自然冷却至室温,减压蒸馏除去溶剂得粗产物,将粗产物过200-300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油醚的体积比1∶7,从而得到为黄色固体的目标产物2-乙烯基异噁唑烷衍生物,产率为88%,纯度为98.3%;
51.熔点:129-156℃;
52.核磁共振:1hnmr(400mhz,氘代氯仿cdcl3)δ7.73-7.82(m,4h),7.47-7.53(m,2h),7.40(t,j=8.4hz,3h),6.92(d,j=5.6hz,2h),5.65(s,1h),5.56(s,1h),4.48(t,j=8.2hz,2h),3.76(t,j=8.2hz,2h),2.30(s,3h);
53.13
c nmr(100mhz,cdcl3)δ160.1,142.5,141.1,139.4,133.6,133.1,131.7,129.1(2c),128.4,128.1,127.8,126.8,126.6,126.22(2c),126.18,124.2,111.8,67.5,34.6,21.5。
54.实施例3
55.向乙腈中,加入o-丙炔基肟醚化合物和磺酰基叠氮、碘化亚铜、三乙胺,然后升温至40℃,并在该温度下搅拌密封反应5小时;其中,o-丙炔基肟醚化合物与碘化亚铜的摩尔比为1∶0.2;o-丙炔基肟醚化合物与三乙胺的摩尔比为1∶1.0;o-丙炔基肟醚化合物与磺酰基叠氮的摩尔比为1∶1.5;以及以毫摩尔计的o-丙炔基肟醚化合物与以毫升计的乙腈的比为1∶9;
56.反应完全后,将反应体系自然冷却至室温,减压蒸馏除去溶剂得粗产物,将粗产物过200-300目硅胶柱色谱层析,以乙酸乙酯和石油醚混合液为洗脱剂,其中乙酸乙酯与石油
醚的体积比1∶5,从而得到为浅黄色液体的目标产物2-乙烯基异噁唑烷衍生物,产率为85%,纯度为96.8%;
57.核磁共振:1hnmr(400mhz,氘代氯仿cdcl3)δ7.80(d,j=8.8hz,2h),7.27(d,j=6.4hz,2h),5.28(s,1h),4.80(s,1h),4.35(t,j=7.0hz,2h),3.66(t,j=7.0hz,2h),2.40(d,j=7.6hz,5h),1.35-1.41(m,2h),1.16-1.26(m,4h),0.84(t,j=5.8hz,3h);
58.13
c nmr(100mhz,cdcl3)δ158.5,142.8,142.1,139.9,129.4(2c),126.6(2c),105.6,67.2,35.0,32.8,31.2,27.6,22.5,21.6,14.1。
59.将实施例1-3的铜催化剂碘化亚铜替换为相同摩尔量的其他铜催化剂,其他操作相同,得到的产物收率分别如下:铜催化剂为氯化亚铜,产物收率为91;铜催化剂为溴化铜,产物收率为82;铜催化剂为三氟甲磺酸铜,产物收率为65;
60.由此可见,在不同的铜催化剂催化下,均能得到相应的产物,总体上一价铜化合物的反应效果比二价的要好,且其中cui对的催化效果最好。
61.将实施例1-3的碱三乙胺替换为相同摩尔量的其他碱,其他操作相同,得到的产物收率分别如下:碱为三乙胺,产物收率为92;碱为三叔丁胺,产物收率为90;碱为2-氟吡啶,产物收率为82;由此可见,在碱中,与三乙胺非常类似的三级胺反应效果也比较好,吡啶类的碱也能得到一定的产物,无机碱的产率则显著的降低。
62.将实施例1-3的有机溶剂乙腈替换为相同体积的其他溶剂,其他操作相同,得到的产物收率分别如下:有机溶剂为meoh,产物收率为痕量;有机溶剂为thf,产物收率为87;有机溶剂为dmf,产物收率为76;
63.由此可见,有机溶剂同样对最终结果有着一定的影响,其中乙腈具有最好的效果,thf次之,其他溶剂的产率都有大幅度的降低。
64.综上所述,由上述所有实施例可明确看出,当采用本发明的方法时,能够使o-丙炔基肟醚化合物和磺酰基叠氮顺利发生反应,从而得到目标产物,且产率良好、后处理简单,这些效果的取得,依赖于多个因素如催化剂、配体、溶剂的综合协同作用。
65.以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为所述的具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1