一种抑制程序性细胞坏死的苯并噻唑衍生物及其应用
1.本发明属于医药技术领域,具体涉及一种抑制程序性细胞坏死的苯并噻唑衍生物及其应用。
背景技术:
2.细胞作为生命体的基本单位,其生存与死亡对于生物个体的发育、进化以及组织器官的生理代谢和平衡起着重要的作用。细胞死亡可分为细胞凋亡和细胞坏死。细胞坏死长期以来都被认为是一种被动且不可调控的过程,然而近几年的科学研究表明,细胞坏死也是受到精密调控的,即程序性细胞坏死。且炎症、肿瘤、心血管疾病、自身免疫性疾病、神经退行性疾病以及代谢性疾病等都与程序性坏死有关。
3.程序性细胞坏死主要由肿瘤坏死因子受体(tnfr)家族或toll样受体(tlr)家族调控启动。死亡受体被激活之后,与受体蛋白相互作用的两个蛋白激酶ripk1和ripk3被激活形成复合体,进而招募ripk3的底物mlkl并催化它发生磷酸化,磷酸化的mlkl发生寡聚化转位到质膜上,引起膜通透性的改变,最终实现坏死的发生。
4.针对这三个靶点,已经开发了几种新型抑制剂。袁钧英团队在2005年首次鉴定出ripk1抑制剂nec-1,随后开发出具有更好药代动力学特性的nec-1s。随后ripk1特异性抑制剂已被开发出来,以苯并恶唑酮(如gsk’772,dnl758)和二氢吡唑(如gsk’547)为代表,作为抗炎剂和神经退行性变疗法。目前以ripk1为靶点的抑制剂数量较多、专一性好;但存在代谢稳定性差、药效低、药代动力学差等问题。ripk3在下游的细胞坏死信号通路中起中心作用,并且可以独立于ripk1发出信号,其除了具有激酶功能,还具有支架蛋白功能,但单纯抑制ripk3的激酶活性反而诱导凋亡。mlkl的抑制剂存在数目少且多显示出选择性不强、细胞毒性高等问题,目前暂无上市的抗坏死药物。同时作用于ripk1和ripk3双靶点的小分子抑制剂报道较少。因此,聚焦ripk1和ripk3双靶点激酶抑制剂的研究,开发高效、低毒的全新小分子尤为必要。
技术实现要素:
5.本发明的第一个目的是提供一种抑制程序性细胞坏死的苯并噻唑衍生物。
6.本发明的第二个目的是提供一种所述苯并噻唑衍生物在制备ripk1、ripk3双靶点激酶抑制剂中的应用。
7.本发明的第三个目的是提供一种所述苯并噻唑衍生物在制备程序性细胞坏死抑制剂中的应用。
8.本发明的第四个目的是提供一种所述苯并噻唑衍生物在制备治疗程序性细胞坏死相关疾病的药物中的应用,或在制备治疗由ripk1和ripk3激酶紊乱、过度激活或过度相互作用引起的疾病的药物中的应用。
9.为了实现上述目的,本发明采用的技术方案如下:
10.本发明的第一方面提供了一种苯并噻唑衍生物或其药用盐,结构如通式i所示:
[0011][0012]
其中,
[0013]
r1选自氢、卤素(氟、氯、溴、碘)、c1~c6烷基、c1~c6烷氧基、硝基、氰基、三氟甲基、苯基;
[0014]
r2选自氢、卤素(氟、氯、溴、碘)、c1~c6烷基、c1~c6烷氧基、硝基、氰基、三氟甲基、苯基;
[0015]
r3选自氢、卤素(氟、氯、溴、碘)、c1~c6烷基、c1~c6烷氧基、硝基、氰基、三氟甲基、苯基;
[0016]
r4选自氢、卤素(氟、氯、溴、碘)、c1~c6烷基、c1~c6烷氧基、硝基、氰基、三氟甲基、苯基;
[0017]
r5选自氢、卤素(氟、氯、溴、碘)、c1~c6烷基、c1~c6烷氧基、硝基、氰基、三氟甲基、苯基。
[0018]
较优选的,所述苯并噻唑衍生物中,
[0019]
r1选自氢、卤素(氟、氯、溴、碘)、甲基、乙基、异丙基、正丙基、叔丁基、甲氧基、乙氧基、异丙氧基、正丙氧基、叔丁氧基、硝基、氰基、三氟甲基、苯基;
[0020]
r2选自氢、卤素(氟、氯、溴、碘)、甲基、乙基、异丙基、正丙基、叔丁基、甲氧基、乙氧基、异丙氧基、正丙氧基、叔丁氧基、硝基、氰基、三氟甲基、苯基;
[0021]
r3选自氢、卤素(氟、氯、溴、碘)、甲基、乙基、异丙基、正丙基、叔丁基、甲氧基、乙氧基、异丙氧基、正丙氧基、叔丁氧基、硝基、氰基、三氟甲基、苯基;
[0022]
r4选自氢、卤素(氟、氯、溴、碘)、甲基、乙基、异丙基、正丙基、叔丁基、甲氧基、乙氧基、异丙氧基、正丙氧基、叔丁氧基、硝基、氰基、三氟甲基、苯基;
[0023]
r5选自氢、卤素(氟、氯、溴、碘)、甲基、乙基、异丙基、正丙基、叔丁基、甲氧基、乙氧基、异丙氧基、正丙氧基、叔丁氧基、硝基、氰基、三氟甲基、苯基。
[0024]
最优选的,所述苯并噻唑衍生物选自以下化合物的一种:
[0025]
[0026]
[0027][0028]
本发明的第二方面提供了一种所述苯并噻唑衍生物或其药用盐在制备ripk1、ripk3双靶点激酶抑制剂中的应用。
[0029]
本发明的第三方面提供了一种所述苯并噻唑衍生物或其药用盐在制备程序性细胞坏死抑制剂中的应用。
[0030]
本发明的第四方面提供了一种所述苯并噻唑衍生物或其药用盐在制备治疗程序性细胞坏死相关疾病的药物中的应用,或在制备治疗由ripk1和ripk3激酶紊乱、过度激活或过度相互作用引起的疾病的药物中的应用。
[0031]
本发明的第五方面提供了一种药物组合物,包括所述苯并噻唑衍生物或其药用盐及药学上可接受的辅料。
[0032]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0033]
本发明的苯并噻唑衍生物可以用于制备程序性细胞坏死相关疾病的防治药物,或用于制备由ripk1和ripk3激酶紊乱、过度激活或过度相互作用引起的疾病的药物中的应用。
[0034]
本发明的全新结构的苯并噻唑衍生物可以作为程序性细胞坏死抑制剂,有效地抑制程序性细胞坏死,达到纳摩尔级,其中活性最好的化合物i-4的抗细胞坏死活性达到6.7纳摩尔,与进入临床研究阶段的gsk’772(抗细胞坏死活性为3.6纳摩尔)相当;特别的是,与现有的临床研究阶段的gsk’772仅作用在细胞坏死上游通路靶点ripk1不同,该类苯并噻唑衍生物可以同时作用在程序性细胞坏死通路的关键靶点ripk1和ripk3上,达到双靶点抑制的效果,并且其激酶亲和力也达到纳摩尔级;可以用于制备程序性细胞坏死相关疾病的防治药物,或用于制备由ripk1、ripk3激酶紊乱、过度激活引起的疾病的药物。
具体实施方式
[0035]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领
域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0036]
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。所有原料未注明合成方法的均购自探索平台、毕得、安耐吉、sigma-aldrich等厂家,均为分析纯。
[0037]
以下化合物的制备从原料到最终产物,没有涉及手性中心的反应。
[0038]
实施例1
[0039]
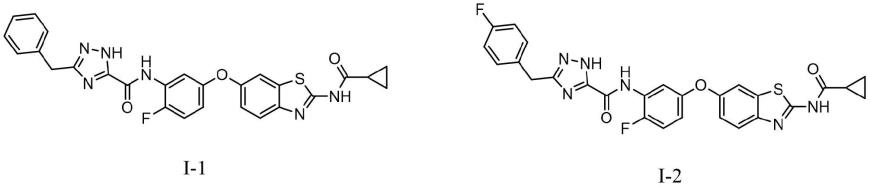
3-苄基-n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-1的制备:
[0040][0041]
步骤a:化合物3的制备
[0042]
将化合物1(1.500g,13.7mmol)、化合物2(1.460ml,13.7mmol)和碳酸钾(5.699g,41.2mmol)加到20ml二甲基亚砜中,85℃加热搅拌2小时,tlc监测反应完全后加水淬灭,乙酸乙酯萃取,饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-10%乙酸乙酯的石油醚溶液),得2.972g白色固体化合物3,收率93.9%。
[0043]
步骤b:化合物4的制备
[0044]
将化合物3(2.892g,12.56mmol)和三氟乙酸酐(3.540ml,25.12mmol)溶于20ml四氢呋喃溶液中,室温搅拌过夜。tlc监测,反应完全后减压浓缩,乙酸乙酯萃取,饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-10%乙酸乙酯的石油醚溶液),得4.006g白色固体化合物4,收率97.63%。
[0045]
步骤c:化合物5的制备
[0046]
将化合物4(4.000g,12.26mmol)溶于20ml乙醇中,将氯化铵溶于5ml水中,然后将氯化铵水溶液加入化合物4的乙醇溶液中,加热至80℃后将铁粉(2.746g,49.04mmol)分批加入,继续搅拌5小时。tlc监测,反应完全后减压浓缩,乙酸乙酯萃取,饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得2.372g白色固体化合物4,收率55.12%。
[0047]
步骤d:化合物6的制备:
[0048]
在室温条件下,将硫氰酸钾(2.490g,25.65mmol)溶于20ml乙酸中,室温搅拌10分钟,再将化合物5(1.900g,6.41mmol)和溴素(431.0μl,9.62mmol)加入到硫氰酸钾的乙酸溶液中,反应液在室温下继续搅拌3小时,tlc监测,反应完全后用水淬灭,饱和碳酸氢钠溶液调至中性,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得2.080g白色固体化合物6,收率91.28%。
[0049]
步骤e:化合物7的制备:
[0050]
在室温条件下,将化合物6(1.555g,4.40mmol)溶于20ml超干吡啶中,氮气保护下0℃搅拌10分钟,再将环丙基甲酰氯(799.0μl,8.80mmol)滴加到化合物6的吡啶溶液中,反应液在0℃下继续搅拌1小时后室温搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得1.267g白色固体化合物7,收率68.31%。
[0051]
步骤f:化合物8的制备:
[0052]
在室温条件下,将化合物7(0.688g,1.63mmol)溶于10ml乙醇中,在0℃下将硼氢化钠(1.235g,32.65mmol)分批加入,反应体系在0℃下继续搅拌1小时后室温搅拌1小时,tlc监测,反应完全后用减压浓缩,乙酸乙酯稀萃取,饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.460g白色固体化合物8,收率86.61%。
[0053]
步骤g:目标化合物i-1的制备:
[0054]
在室温条件下,将化合物8(0.200g,0.58mmol)、化合物9(该化合物制备参考文献:cn105121432a)(0.237g,1.16mmol)和2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(0.441g,1.16mmol)溶于5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.189g白色固体化合物i-1,收率61.76%。1h nmr(500mhz,dmso-d6)δ14.58(s,1h),12.66(s,1h),9.96(s,1h),7.75-7.73(d,j=8.7hz,1h),7.70-7.69(d,j=2.5hz,1h),7.55-7.54(dd,j=6.5,3.1hz,1h),7.33-7.32(s,1h),7.31(s,1h),7.30-7.29(d,j=3.8hz,3h),7.25-7.22(m,1h),7.17-7.14(dd,j=8.7,2.5hz,1h),6.89-6.87(m,1h),4.15(s,2h),2.01-1.98(m,1h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
27h21
fn6o3s[m+h]
+
529.1408;found 529.1438.
[0055]
实施例2
[0056]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-氟苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-2的制备:
[0057]
[0058]
化合物10参考化合物9的方法制备获得。
[0059]
在室温条件下,将化合物10(0.166g,0.75mmol)、化合物8(0.125g,0.36mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.285g,0.75mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.073g白色固体化合物i-2,收率43.06%。1h nmr(600mhz,dmso-d6)δ14.81(s,1h),12.62(s,1h),9.94(s,1h),7.75-7.73(d,j=8.8hz,1h),7.69(d,j=2.5hz,1h),7.54(s,1h),7.34-7.30(m,3h),7.17-7.13(m,3h),6.90-6.87(m,1h),4.15(s,2h),2.01-1.97(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
f2n6o3s[m+h]
+
547.1308;found 547.1336.
[0060]
实施例3
[0061]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-氟苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-3的制备:
[0062][0063]
化合物11参考化合物9的方法制备获得。
[0064]
在室温条件下,将化合物11(0.257g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.185g白色固体化合物i-3,收率58.54%。1h nmr(600mhz,dmso-d6)δ14.78(s,1h),12.59(s,1h),9.93(s,1h),7.71-7.69(d,j=8.8hz,1h),7.65(d,j=2.5hz,1h),7.49-7.48(d,j=6.5hz,1h),7.34-7.31(m,1h),7.29-7.26(t,j=9.6hz,1h),7.12-7.08(m,3h),7.05-7.02(m,1h),6.86-6.83(m,1h),4.14(s,2h),1.97-1.93(m,1h),0.92-0.89(t,j=5.7hz,4h).hrms(esi,positive)m/z calcd for c
27h20
f2n6o3s[m+h]
+
547.1308;found 547.1336.
[0065]
实施例4
[0066]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-氟苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-4的制备:
[0067][0068]
化合物12参考化合物9的方法制备获得。
[0069]
在室温条件下,将化合物12(0.257g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干
吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥,过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.123g白色固体化合物i-4,收率38.92%。1h nmr(600mhz,dmso-d6)δ14.54(s,1h),12.63(s,1h),9.85(s,1h),7.75-7.73(d,j=8.8hz,1h),7.69-7.68(d,j=2.5hz,1h),7.54(s,1h),7.37-7.29(m,3h),7.21-7.14(m,3h),6.90-6.87(m,1h),4.19(s,2h),2.00-1.97(m,1h),0.96-0.94(t,j=5.4hz 4h).hrms(esi,positive)m/z calcd for c
27h20
f2n6o3s[m+h]
+
547.1308;found547.1340.
[0070]
实施例5
[0071]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-氯苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-5的制备:
[0072][0073]
化合物13参考化合物9的方法制备获得。
[0074]
在室温条件下,将化合物13(0.276g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.133g白色固体化合物i-5,收率40.73%。1h nmr(600mhz,dmso-d6)δ14.65(s,1h),12.63(s,1h),9.96(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.52(s,1h),7.39-7.38(m,2h),7.33-7.30(m,3h),7.16-7.14(m,1h),6.90-6.87(m,1h),4.16(s,2h),1.99-1.98(m,1h),0.95-0.94(t,j=5.8hz,4h).hrms(esi,positive)m/z calcd for c
27h20
clfn6o3s[m+h]
+
563.1008;found 563.1046.
[0075]
实施例6
[0076]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-氯苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-6的制备:
[0077][0078]
化合物14参考化合物9的方法制备获得。
[0079]
在室温条件下,将化合物14(0.276g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物
通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.150g白色固体化合物i-6,收率46.01%。1h nmr(600mhz,dmso-d6)δ14.51(s,1h),12.63(s,1h),9.87(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.54(s,1h),7.38-7.30(m,4h),7.26-7.24(dd,j=7.5,1.5hz,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.90-6.87(dd,j=8.5,4.0hz,1h),4.18(s,2h),2.01-1.98(m,1h),0.97-0.93(t,j=5.8hz,2h).hrms(esi,positive)m/z calcd for c
27h20
clfn6o3s[m+h]
+
563.1008;found 563.1042.
[0080]
实施例7
[0081]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-氯苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-7的制备:
[0082][0083]
化合物15参考化合物9的方法制备获得。
[0084]
在室温条件下,将化合物15(0.276g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.182g白色固体化合物i-7,收率55.83%。1h nmr(500mhz,dmso-d6)δ14.51(s,1h),12.63(s,1h),9.86(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.4hz,1h),7.53(s,1h),7.47-7.45(m,1h),7.39-7.29(m,4h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.90-6.86(dd,j=8.4,4.1hz,1h),4.27(s,2h),1.99(m,1h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
27h20
clfn6o3s[m+h]
+
563.1008;found 563.1042.
[0085]
实施例8
[0086]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-溴苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-8的制备:
[0087][0088]
化合物16参考化合物9的方法制备获得。
[0089]
在室温条件下,将化合物16(0.327g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.171g白色固体化合物i-8,收率48.57%。1h nmr(500mhz,dmso-d6)δ14.50(s,1h),12.63(s,1h),9.96(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.53-7.51(dd,j=6.5,4.5hz,3h),
7.31-7.29(t,j=9.6hz,1h),7.26-7.24(m,2h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.90-6.87(m,1h),4.14(s,2h),1.99(m,1h),0.97-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
brfn6o3s[m+h]
+
607.0508;found 607.0536.
[0090]
实施例9
[0091]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-溴苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-9的制备:
[0092][0093]
化合物17参考化合物9的方法制备获得。
[0094]
在室温条件下,将化合物17(0.327g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.213g白色固体化合物i-9,收率60.51%。1h nmr(500mhz,dmso-d6)δ14.49(s,1h),12.63(s,1h),9.85(s,1h),7.75-7.73(d,j=8.8hz,1h),7.69(d,j=2.6hz,1h),7.59-7.45(t,j=29.7hz,3h),7.33-7.29(m,3h),7.16-7.14(dd,j=8.8,2.6hz,1h),6.88(s,1h),4.19(s,2h),2.01-1.98(m,1h),0.96-0.92(m,4h).hrms(esi,positive)m/z calcd for c
27h20
brfn6o3s[m+h]
+
607.0508;found 607.0556.
[0095]
实施例10
[0096]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-溴苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-10的制备:
[0097][0098]
化合物18参考化合物9的方法制备获得。
[0099]
在室温条件下,将化合物18(0.327g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.252g白色固体化合物i-10,收率71.19%。1h nmr(600mhz,dmso-d6)δ14.48(s,1h),12.62(s,1h),9.83(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.64-7.63(d,j=8.0hz,1h),7.54-7.46(m,1h),7.38-7.30(m,3h),7.23(s,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.89(s,1h),4.27(s,2h),2.01-1.97(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
brfn6o3s[m+h]
+
607.0508;found 607.0553.
[0100]
实施例11
[0101]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-碘苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-11的制备:
[0102][0103]
化合物19参考化合物9的方法制备获得。
[0104]
在室温条件下,将化合物19(0.382g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.246g白色固体化合物i-11,收率64.91%。1h nmr(500mhz,dmso-d6)δ14.52(s,1h),12.59(s,1h),9.89(s,1h),7.71-7.69(d,j=8.7hz,1h),7.65-7.63(m,3h),7.49(s,1h),7.29-7.26(t,j=9.6hz,1h),7.12-7.05(m,3h),6.86-6.83(m,1h),4.08(s,2h),1.97-1.94(m,1h),0.93-0.90(m,4h).hrms(esi,positive)m/z calcd for c
27h20
ifn6o3s[m+h]
+
655.0308;found 655.0407.
[0105]
实施例12
[0106]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-碘苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-12的制备:
[0107][0108]
化合物20参考化合物9的方法制备获得。
[0109]
在室温条件下,将化合物20(0.382g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.229g白色固体化合物i-12,收率61.07%。1h nmr(500mhz,dmso-d6)δ14.49(s,1h),12.63(s,1h),9.92(s,1h),7.75-7.73(d,j=8.8hz,1h),7.69(d,j=2.6hz,2h),7.63-7.61(d,j=7.9hz,1h),7.52(s,1h),7.34-7.29(m,2h),7.16-7.12(m,2h),6.90-6.88(dd,j=9.0,3.6hz,1h),4.13(s,2h),2.00-1.97(m,1h),0.97-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
ifn6o3s[m+h]
+
655.0308;found 655.0406.
[0110]
实施例13
[0111]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-碘苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-13的制备:
[0112][0113]
化合物21参考化合物9的方法制备获得。
[0114]
在室温条件下,将化合物21(0.382g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.203g白色固体化合物i-13,收率54.13%。1h nmr(600mhz,dmso-d6)δ14.47(s,1h),12.63(s,1h),9.87(s,1h),7.89-7.87(m,1h),7.75-7.73(d,j=8.7hz,1h),7.70-7.69(d,j=2.5hz,1h),7.52(s,1h),7.39-7.37(t,j=7.5hz,1h),7.33-7.29(m,2h),7.16-7.14(dd,j=8.7,2.5hz,1h),7.05-7.03(t,j=7.6hz,1h),6.89-6.88(d,j=8.8hz,1h),4.25(s,2h),2.00-1.98(m,1h),0.97-0.92(m,4h).hrms(esi,positive)m/z calcd for c
27h20
ifn6o3s[m+h]
+
655.0308;found 655.0413.
[0115]
实施例14
[0116]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-甲基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-14的制备:
[0117][0118]
化合物22参考化合物9的方法制备获得。
[0119]
在室温条件下,将化合物22(0.252g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.115g白色固体化合物i-14,收率36.86%。1h nmr(600mhz,dmso-d6)δ14.52(s,1h),12.63(s,1h),10.05-9.91(d,j=84.9hz,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.54(s,1h),7.33-7.30(t,j=9.6hz,1h),7.17-7.14(m,3h),7.13-7.11(d,j=7.9hz,2h),6.89-6.86(m,1h),4.09(s,2h),2.25(s,3h),1.99-1.97(m,1h),0.97-0.93(m,4h).hrms(esi,positive)m/zcalcd for c
28h23
fn6o3s[m+h]
+
543.1508;found 543.1611.
[0120]
实施例15
[0121]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-甲基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-15的制备:
[0122][0123]
化合物23参考化合物9的方法制备获得。
[0124]
在室温条件下,将化合物23(0.252g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.143g白色固体化合物i-15,收率45.54%。1h nmr(500mhz,dmso-d6)δ14.51(s,1h),12.65(s,1h),9.85(s,1h),7.75-7.73(d,j=8.8hz,1h),7.69-7.68(d,j=2.5hz,1h),7.60(s,1h),7.31-7.29(t,j=9.6hz,1h),7.21-7.14(m,2h),7.09-7.03(m,3h),6.89-6.86(dd,j=8.2,4.2hz,1h),4.11(s,2h),2.26(s,3h),2.02-2.00(m,1h),0.95-0.94(d,j=6.2hz,4h).hrms(esi,positive)m/z calcd for c
28h23
fn6o3s[m+h]
+
543.1508;found 543.1611.
[0125]
实施例16
[0126]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-硝基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-16的制备:
[0127][0128]
化合物24参考化合物9的方法制备获得。
[0129]
在室温条件下,将化合物24(0.288g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.115g白色固体化合物i-16,收率36.86%。1h nmr(600mhz,dmso-d6)δ14.52(s,1h),12.63(s,1h),10.05-9.91(d,j=84.9hz,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.54(s,1h),7.33-7.30(t,j=9.6hz,1h),7.17-7.14(m,3h),7.13-7.11(d,j=7.9hz,2h),6.89-6.86(m,1h),4.09(s,2h),2.25(s,3h),1.99-1.97(m,1h),0.97-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
fn7o5s[m+h]
+
574.1208;found 574.1298.
[0130]
实施例17
[0131]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-硝基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-17的制备:
[0132][0133]
化合物25参考化合物9的方法制备获得。
[0134]
在室温条件下,将化合物25(0.252g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.110g白色固体化合物i-17,收率33.13%。1h nmr(600mhz,dmso-d6)δ14.56(s,1h),12.62(s,1h),9.90(s,1h),8.21(s,1h),8.14-8.12(m,1h),7.78-7.76(m,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.65-7.62(t,j=7.9hz,1h),7.51(s,1h),7.33-7.30(dd,j=10.2,9.0hz,1h),7.16-7.14(dd,j=8.8,2.5hz,1h),6.90-6.88(dd,j=8.5,4.2hz,1h),4.34(s,2h),2.00-1.98(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
27h20
fn7o5s[m+h]
+
574.1208;found 574.1324.
[0135]
实施例18
[0136]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-硝基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-18的制备:
[0137][0138]
化合物26参考化合物9的方法制备获得。
[0139]
在室温条件下,将化合物26(0.252g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.178g白色固体化合物i-18,收率53.61%。1h nmr(600mhz,dmso-d6)δ14.46(s,1h),12.62(s,1h),9.90-9.77(m,1h),8.08-8.07(d,j=8.2hz,1h),7.75-7.73(d,j=8.7hz,2h),7.69-7.68(d,j=2.5hz,1h),7.60-7.59(m,3h),7.33-7.30(dd,j=10.2,9.0hz,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.89-6.87(d,j=9.0hz,1h),4.50(s,2h),2.01-1.97(m,1h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
27h20
fn7o5s[m+h]
+
574.1208;found 574.1304.
[0140]
实施例19
[0141]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-三氟甲基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-19的制备:
[0142][0143]
化合物27参考化合物9的方法制备获得。
[0144]
在室温条件下,将化合物27(0.315g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.194g白色固体化合物i-19,收率56.07%。1h nmr(600mhz,dmso-d6)δ14.56(s,1h),12.62(s,1h),9.96(s,1h),7.75-7.73(m,1h),7.70-7.68(m,3h),7.52-7.51(m,3h),7.33-7.29(dd,j=10.2,9.0hz,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.89-6.88(m,1h),4.27(s,2h),2.01-1.98(m,1h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
28h20
f4n6o3s[m+h]
+
597.1308;found597.1329.
[0145]
实施例20
[0146]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-三氟甲基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-20的制备:
[0147][0148]
化合物28参考化合物9的方法制备获得。
[0149]
在室温条件下,将化合物28(0.315g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.213g白色固体化合物i-20,收率61.56%。1h nmr(500mhz,dmso-d6)δ14.56(s,1h),12.63(s,1h),9.89(s,1h),7.75-7.73(d,j=8.7hz,1h),7.70-7.69(m,2h),7.61-7.56(m,4h),7.33-7.29(dd,j=10.2,9.0hz,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.90-6.87(m,1h),4.29(s,2h),2.01-1.98(m,1h),0.96-0.94(m,j=7.9,2.4hz,4h).hrms(esi,positive)m/z calcd for c
28h20
f4n6o3s[m+h]
+
597.1308;found 597.1327.
[0150]
实施例21
[0151]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-三氟甲基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-21的制备:
[0152][0153]
化合物29参考化合物9的方法制备获得。
[0154]
在室温条件下,将化合物29(0.315g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.250g白色固体化合物i-21,收率72.25%。1h nmr(600mhz,dmso-d6)δ14.49(s,1h),12.63(s,1h),9.84(s,1h),7.75-7.73(d,j=8.8hz,2h),7.69(d,j=2.5hz,1h),7.65-7.64(d,j=7.6hz,1h),7.50-7.45(m,3h),7.33-7.30(t,j=9.6hz,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.89(s,1h),4.34(s,2h),2.00-1.98(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
28h20
f4n6o3s[m+h]
+
597.1308;found 597.1323.
[0155]
实施例22
[0156]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-甲氧基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-22的制备:
[0157][0158]
化合物30参考化合物9的方法制备获得。
[0159]
在室温条件下,将化合物30(0.271g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.139g白色固体化合物i-22,收率42.90%。1h nmr(600mhz,dmso-d6)δ14.70(s,1h),12.66(s,1h),9.85(s,1h),7.75-7.73(d,j=8.7hz,1h),7.68(d,j=2.5hz,1h),7.56(s,1h),7.31-7.29(dd,j=10.2,9.0hz,1h),7.22-7.21(d,j=8.2hz,2h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.88-6.87(m,3h),4.08(s,2h),3.71(s,3h),2.04-2.02(m,1h),0.95-0.94(m,4h).hrms(esi,positive)m/z calcd for c
28h23
fn6o4s[m+h]
+
559.1508;found 559.1534.
[0160]
实施例23
[0161]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-甲氧基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-23的制备:
[0162][0163]
化合物31参考化合物9的方法制备获得。
[0164]
在室温条件下,将化合物31(0.271g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.112g白色固体化合物i-23,收率34.57%。1h nmr(600mhz,dmso-d6)δ14.47(m,1h),12.63(s,1h),9.84(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.60-7.59(d,j=6.1hz,1h),7.33-7.30(t,j=9.6hz,1h),7.23(s,1h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.88-6.83(m,4h),4.14(s,2h),3.73(s,3h),2.00-1.98(m,1h),0.96-0.94(dd,j=6.3,2.9hz,4h).hrms(esi,positive)m/z calcd for c
28h23
fn6o4s[m+h]
+
559.1508;found 559.1549.
[0165]
实施例24
[0166]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(2-甲氧基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-24的制备:
[0167][0168]
化合物32参考化合物9的方法制备获得。
[0169]
在室温条件下,将化合物32(0.271g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.171g白色固体化合物i-24,收率52.62%。1h nmr(600mhz,dmso-d6)δ14.47(m,1h),12.64(s,1h),9.84(s,1h),7.75-7.73(d,j=8.7hz,1h),7.68(d,j=2.5hz,1h),7.55(s,1h),7.32-7.29(dd,j=10.2,9.0hz,1h),7.27-7.24(m,1h),7.16-7.14(m,2h),7.00-6.98(dd,j=8.3,1.1hz,1h),6.91-6.85(m,2h),4.08(s,2h),3.77(s,3h),2.02-2.00(t,j=5.0hz,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
28h23
fn6o4s[m+h]
+
559.1508;found 559.1583.
[0170]
实施例25
[0171]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-氰基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-25的制备:
[0172][0173]
化合物33参考化合物9的方法制备获得。
[0174]
在室温条件下,将化合物33(0.265g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.163g白色固体化合物i-25,收率50.13%。1h nmr(600mhz,dmso-d6)δ14.52(m,1h),12.64(s,1h),9.99(s,1h),7.80-7.79(d,j=8.4hz,2h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.50-7.49(m,3h),7.33-7.30(dd,j=10.2,9.0hz,1h),7.16-7.14(dd,j=8.7,2.6hz,1h),6.90-6.87(dt,j=8.9,3.3hz,1h),4.27(s,2h),2.00-1.98(dt,j=7.4,2.9hz,1h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
28h20
fn7o3s[m+h]
+
554.1308;found 554.1379.
[0175]
实施例26
[0176]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(3-氰基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-26的制备:
[0177][0178]
化合物34参考化合物9的方法制备获得。
[0179]
在室温条件下,将化合物34(0.265g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.188g白色固体化合物i-26,收率57.8%。1h nmr(600mhz,dmso-d6)δ14.62(s,1h),12.63(s,1h),9.94(s,1h),7.79(s,1h),7.75-7.73(d,j=8.8hz,2h),7.69(d,j=2.5hz,1h),7.65-7.64(d,j=7.8hz,1h),7.56-7.53(t,j=7.8hz,2h),7.33-7.30(t,j=9.6hz,1h),7.16-7.14(dd,j=8.7,2.6hz,1h),7.90-6.88(dd,j=8.4,4.2hz,1h),4.24(s,2h),1.99(m,1h),0.96-0.93(t,j=5.4hz,4h).hrms(esi,positive)m/z calcd for c
28h20
fn7o3s[m+h]
+
554.1308;found 554.1383.
[0180]
实施例27
[0181]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-异丙基基苄基)-1h-1,2,4-三唑-3-甲酰胺即化合物i-27的制备:
[0182][0183]
化合物35参考化合物9的方法制备获得。
[0184]
在室温条件下,将化合物35(0.285g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.155g白色固体化合物i-27,收率46.97%。1h nmr(500mhz,dmso-d6)δ14.61(s,1h),12.65(s,1h),9.89-9.86(m,1h),7.75-7.69(m,2h),7.55(s,1h),7.33-7.29(t,j=9.6hz,1h),7.19(s,5h),6.88-6.86(m,1h),4.10(s,2h),2.84-2.82(m,1h),2.02-1.98(m,1h),1.16-1.15(d,j=7.0hz,6h),0.96-0.94(m,4h).hrms(esi,positive)m/z calcd for c
30h27
fn6o3s[m+h]
+
571.1808;found 571.189.
[0185]
实施例28
[0186]
n-(5-((2-(环丙甲酰氨基)-苯并[d]噻唑-6-基)-氧)-2-氟苯基)-3-(4-乙基联苯)-1h-1,2,4-三唑-3-甲酰胺即化合物i-28的制备:
[0187][0188]
化合物36参考化合物9的方法制备获得。
[0189]
在室温条件下,将化合物36(0.324g,1.16mmol)、化合物8(0.200g,0.58mmol)和2-(7-氮杂苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(0.441g,1.16mmol)加入到5ml超干吡啶中,反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-30%乙酸乙酯的石油醚溶液),得0.266g白色固体化合物i-28,收率75.78%。1h nmr(600mhz,dmso-d6)δ14.53(s,1h),12.63(s,1h),9.95(s,1h),7.75-7.73(d,j=8.7hz,1h),7.69(d,j=2.5hz,1h),7.63-7.61(m,4h),7.55(s,1h),7.45-7.43(t,j=7.8hz,2h),7.38-7.37(d,j=8.2hz,2h),7.35-7.30(m,2h),7.16-7.14(dd,j=8.7,2.5hz,1h),6.89-6.87(m,1h),4.20(s,2h),2.01-1.98(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
33h25
fn6o3s[m+na]
+
627.1508;found 627.1612.
[0190]
对比例1
[0191]
szm-594的制备
[0192]
(s)-3-苄基-n-(5-甲基-4-氧代-2,3,4,5-四氢苯并[b][1,4]恶唑啉-3-基)-1h-1,2,4-三唑-5-甲酰胺(37)的制备即化合物szm-594的制备:
[0193]
合成路线如下:
[0194][0195]
步骤a-f按照实施例1化合物i-1的步骤a-f制备。
[0196]
步骤h:目标化合物37的制备:
[0197]
在室温条件下,将化合物8(0.200g,0.58mmol)溶于5ml超干吡啶中,再加入间三氟甲基苯乙酸(0.237mg,1.16mmol),反应体系在85℃氮气保护下搅拌过夜,tlc监测,反应完全后用水淬灭,乙酸乙酯萃取,并用饱和氯化钠溶液洗涤3次,无水硫酸钠干燥、过滤,减压下浓缩得到粗产物,粗产物通过硅胶柱色谱纯化(洗脱剂:含0-50%乙酸乙酯的石油醚溶液),得0.166g白色固体化合物37,收率54.25%。1h nmr(600mhz,dmso-d6)δ12.61(s,1h),10.11(s,1h),7.72-7.67(m,3h),7.63(d,j=2.4hz,1h),7.61-7.59(m,2h),7.56-7.53(m,1h),7.29-7.26(m,1h),7.10(dd,j=2.8hz,8.4hz,1h),6.81-6.78(m,1h),3.86(s,2h),2.01-1.97(m,1h),0.96-0.93(m,4h).hrms(esi,positive)m/z calcd for c
26h19
f4n3o3s[m+h]
+
530.1156;found 530.1169.
[0198]
实施例29
[0199]
ripk1、ripk3激酶实验
[0200]
采用kinomescan
tm
的ripk1和ripk3激酶检测方法,测试了本发明实施例1~28合成化合物和部分中间体、对比例1制备的化合物szm-594、阳性对照分子gsk’772对ripk1和ripk3的激酶活性。
[0201]
使用qpcr检测dna所标记ripk1和ripk3激酶-标记t7噬菌体菌株裂解物。将激酶、配体亲和珠和测试化合物在结合缓冲液(20%seablock,0.17
×
pbs,0.05%tween 20,6mm dtt)中结合反应。将化合物3倍稀释的方法,设置10个点,室温下振荡孵育1h,并用洗涤缓冲液(1
×
pbs,0.05%tween 20)洗涤亲和珠,然后将亲和珠在洗脱缓冲液(1
×
pbs,0.05%吐温20,0.5μm非生物素化的亲和配体)中重悬,并在室温下振荡孵育30min。通过qpcr测量洗脱液中的激酶浓度,计算kd值,计算公式如下(hill slope设置为-1),结果见表1:
[0202][0203]
实施例30
[0204]
抗程序性细胞坏死实验
[0205]
本发明实施例1~28,对比例1制备的化合物szm-594以及阳性对照分子gsk’772的抗程序性细胞坏死活性测试。通过z-vad-fmk(20mm)和smac模拟物(10μm)预处理30分钟后,
加入htnf-α(20ng/ml)诱导肿瘤细胞(ht-29,人结肠癌细胞)的坏死。将本发明实施例1~28、对比例1制备的化合物szm-594以及阳性对照分子gsk’772分别与上述组合的细胞一起孵育10小时。使用celltiter-glo发光细胞活力测定试剂盒(promega)测试细胞活性。用spectramax m5酶标仪(molecular devices,california)记录化学发光值,计算ec
50
值,结果见表1。
[0206]
表1.化合物的抗程序性细胞坏死活性
[0207]
[0208][0209]
从表1可以看出,本发明提供了全新的苯并噻唑衍生物结构,大部分化合物对于htnf-α诱导的程序性细胞坏死具有较好的抑制活性,达到了纳摩尔级,其中化合物i-4的细胞活性明显优于对比例1制备的化合物szm-594,与进入临床研究阶段的gsk’772相当,也明显优于化合物szm630。针对激酶ripk1和ripk3,化合物的活性均达到了纳摩尔级(ripk1 kd《10nm,ripk3 kd《100nm),也均优于对比例1制备的化合物szm-594。然而现有的临床研究阶段的gsk’772仅作用在ripk1,对ripk3无效;szm630仅作用在ripk3,对ripk1无效;此外,本发明的化合物对ripk1的抑制活性也优于gsk’772,且对ripk3的抑制剂活性呈现出纳摩尔级。因此,本发明的新型苯并噻唑衍生物可以作为程序性细胞坏死抑制剂,有效地抑制程序性细胞坏死;作用靶点ripk1和ripk3激酶,可以用于制备程序性细胞坏死相关疾病的防治药物,或制备由ripk1、ripk3激酶紊乱、过度激活引起的疾病的药物中的应用。
[0210]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1