一种二胺、液晶取向剂及其制备方法和应用与流程
1.本技术涉及一种二胺、液晶取向剂及其制备方法和应用,属于液晶取向剂领域。
背景技术:
2.液晶显示器(lcd)在笔记本电脑、监视器、液晶电视等领域有着广泛的应用。在lcd产品制造中,提高良品率,增加产品竞争力是lcd厂家一直追求的目标。然而在lcd制造中,经常会出现都画面闪烁、封口污染、液晶冲刷纹、显示不匀、摩擦痕迹等不良问题。
3.为了解决上述问题,需要从lcd制造工艺和原材料两方面改善。在液晶显示器中,液晶取向膜对液晶分子的取向取向控制十分重要,其优劣直接影响液晶显示器件的品质。通常液晶显示器电压保持率低(vhr)、蓄积电荷(rdc)高的情况下,驱动后无法正常控制液晶分子取向,导致显示对比度降低,画面闪烁、封口污染等问题,显著降低液晶显示器件的显示品质。若预倾角稳定性差,则显示器易出现灌注液晶过程中产生的液晶冲刷纹,同时可能出现显示不匀等问题。
4.对于聚酰亚胺的液晶取向膜,为了获得电学性能优良的液晶取向剂,专利jp2013152421a和专利jpwo20151224中分别公开了一种含氮六元环的二胺结构,用于制备可溶性聚酰亚胺的液晶取向剂;专利 jph09316200a中公开了一种含有特定结构的叔胺的液晶取向剂。
5.另外,为了提高液晶取向膜对液晶的较低预倾角及预倾角稳定性,专利jph10123532a中公开了含有特定二胺的聚酰胺酸液晶取向剂。专利 wo2015156335中通过具有烷氧烷及脲基的化合物获得了电压保持率高、液晶取向性优异且蓄积电荷少的液晶取向剂。
6.苯并环丁烯是一种活性化合物,在200以上环丁烯环可进行开环聚合反应,具有优异的绝缘性。专利cn111764002a通过酰胺键连接苯并环丁烯,制备了一种侧链为苯并环丁烯的二胺,公开了一种低介电常数的聚酰亚胺纤维的制备方法;专利cn113336998a中制备了一种苯并环丁烯交联型的具有微孔的聚酰亚胺薄膜,该膜具有较低的介电常数和导热系数;专利cn113773432a中公开了一种侧基为苯并环丁烯的低介电常数聚苯乙烯材料。
技术实现要素:
7.根据本技术的第一个方面,提供了一种二胺化合物。该二胺化合物为一种新型的含苯并环丁烯的二胺,该二胺化合物结构中通过醚键与苯并环丁烯连接,相对于现有技术中的采用酰胺键将苯并环丁烯连接到二胺上,醚键不易水解,储存稳定性更好。由于苯并环丁烯在200以上加热,环丁烯结构发生开环反应,形成八元环结构,阻断苯环间电荷传输,提高电绝缘性。通过醚键连接的苯并环丁烯侧基具有更好的柔性,可降低苯并环丁烯交联时的空间阻力,保证了聚合物的交联程度,从而使聚酰亚胺取向膜具有较低的介电常数。同时苯并环丁烯间的交联作用,进一步使聚酰亚胺薄膜的力学性能,耐摩擦性能明显提高。
8.本技术通过深入的研究,通过液晶取向剂中关键成分的选择,解决了背景技术中
提到的难题,即通过本发明的液晶取向剂,形成液晶取向膜,机械性能优异,电绝缘性佳,在实施摩擦处理时不易因摩擦处理而产生摩擦斜纹,耐摩擦性好、通过本发明液晶取向剂所制液晶显示器件的vhr高、rdc低、预倾角稳定性高、功耗低、可靠性高、有效提高 lcd的良品率。
9.一种二胺化合物,具有通式(1)所示结构:
[0010][0011]
其中,ar选自苯基、萘基、联苯基、二苯醚基中的一种;
[0012]
r1选自氢原子、甲基、乙基、叔丁基、叔戊基中的一种;
[0013]
r2具有式(2)所示结构
[0014][0015]
n为1或2。
[0016]
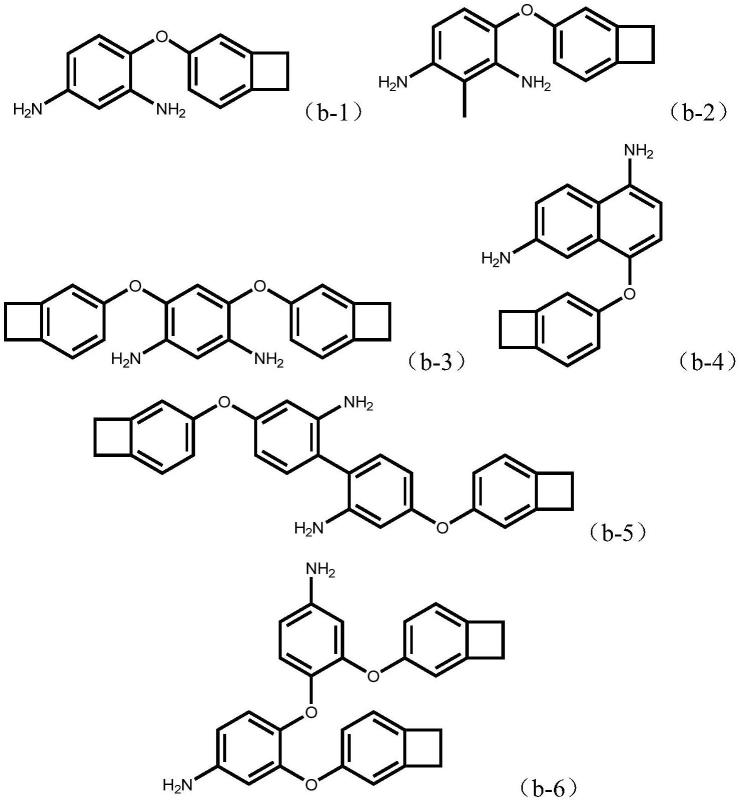
进一步的,从所制液晶取向膜的性能和式(1)所示结构二胺的制备难易程度的角度出发,二胺化合物优选为下述(b-1)-(b-6)所示结构:
[0017][0018]
上述二胺化合物结构中的苯并环丁烯在室温下稳定储存,在200以上加热,环丁烯结构发生开环反应,形成八元环结构,如下所示,该交联结构阻断苯环间电荷传输,降低了分子极化率,从而使聚合物的介电常数明显降低。
[0019][0020]
根据本技术的第二个方面,提供了一种聚酰亚胺前体。
[0021]
一种聚酰亚胺前体,具有通式(3)所示结构:
[0022][0023]
其中,r3为四羧酸二酐的4价有机基团;
[0024]
r4为氢原子或碳原子数1~5的烷基;
[0025]
r5为上述所述的二胺化合物的2价有机基团。
[0026]
可选地,所述四羧酸二酐包括脂肪族四羧酸二酐、芳香族四羧酸二酐中的至少一种。
[0027]
可选地,所述脂肪族四羧酸二酐选自环丁烷四羧酸二酐、1,2,3,4-环戊烷四羧酸二酐、2,3,5-三羧基环戊基乙酸二酐、1,2,4,5-环己烷四羧酸二酐、3,4-二羧基-1,2,3,4-四氢-1-萘琥珀酸二酐、3-羧甲基-1,2,4-环戊烷三甲酸1,4:2,3-双酐中的至少一种;
[0028]
所述芳香族四羧酸二酐选自均苯四酸二酐、3,3’,4,4
’‑
联苯四羧酸二酐、3,3’,4,4
’‑
二苯酮四酸二酐、2,3,3’,4
’‑
二苯酮四酸二酐、双 (3,4-二羧基苯基)醚二酐、双(3,4-二羧基苯基)砜二酐、1,2,5,6-萘四酸二酐、4,4'-(4,4'-异丙基二苯氧基)二酞酸酐、4,4
′‑
(六氟异丙烯)二酞酸酐、4,4'-异亚丙基-双(邻苯二甲酸酐)中的至少一种。
[0029]
为了提高取向膜与基材的粘附性,还可以在聚酰亚胺前体或聚酰亚胺中引入1,3-双(3-氨基丙基)四甲基二硅氧烷、3,3'-(1,4-亚苯基双(二甲基硅烷二基))双(丙-1-胺)、4,4'-(1,1,3,3-四甲基二硅氧烷-1,3-二基)二苯胺中的至少一种二胺单体;优选的,上述二胺单体占二胺摩尔量的不超过5%。
[0030]
衍生自脂肪族的r3占r3总量的20%~100%。更优选的,按物质的量计,所述衍生自脂肪族的r3占r3总量的50%~100%。
[0031]
根据本技术的第三个方面,提供了一种二胺化合物的制备方法。该方法工艺简单,可获得的高纯度的二胺单体。
[0032]
上述所述二胺化合物的制备方法,包括以下步骤:
[0033]
s1、将式(5)所示卤代化合物加入到含有式(4)所示羟基化合物、第一催化剂、第一溶剂的混合物中,反应 得到二硝基化合物;
[0034][0035]
s2、将含有所述二硝基化合物、第二溶剂、第二催化剂、还原剂的混合物,反应 得到二胺化合物;
[0036]
ar、r1、n与上述式(1)中ar、r1、n定义一致;x为卤素原子。
[0037]
可选地,步骤s1中,所述第一催化剂选自氢化钠(60wt%)、氢化钾、氢化钙中的至少一种;
[0038]
所述第一溶剂选自四氢呋喃、1,4-二氧六环、n-甲基吡咯烷酮、n-乙基吡咯烷酮中的至少一种。
[0039]
所述x为氟原子。
[0040]
可选地,步骤s1中,式(4)所示羟基化合物与式(5)所示化合物中的卤原子的摩尔比为1:0.9~1.1。
[0041]
第一催化剂与式(4)所示羟基化合物的摩尔比为1~1.5:1。
[0042]
可选地,步骤s1中,式(4)所示羟基化合物与式(5)所示化合物中的卤原子的摩尔比独立地选自1:0.90、1:0.95、1:1.0、1:1.05、1: 1.10中的任意值或任意两者之间的范围值。
[0043]
可选地,第一催化剂与式(4)所示羟基化合物的摩尔比独立地选自 1.0:1、1.1:1、1.2:1、1.3:1、1.4:1、1.5:1中的任意值或任意两者之间的范围值。
[0044]
可选地,步骤s1中,反应 的条件如下:
[0045]
温度为0~30;
[0046]
时间为2h~24h。
[0047]
可选地,步骤s1中,反应 在非活性气氛下进行;
[0048]
所述非活性气氛选自氮气、氩气中的至少一种。
[0049]
可选地,步骤s2中,所述二硝基化合物与所述第二催化剂的质量比为1:(0.01~0.5);
[0050]
可选地,步骤s2中,所述二硝基化合物与所述第二催化剂的质量比独立地选自1:0.01、1:0.03、1:0.05、1:0.07、1:0.10、1:0.20、1: 0.30、1:0.40、1:0.50中的任意值或任意两者之间的范围值。
[0051]
所述二硝基化合物与所述还原剂的摩尔比为1:1~1.5;
[0052]
所述二硝基化合物与所述第二溶剂的质量比为1:20~200。
[0053]
可选地,所述二硝基化合物与所述第二溶剂的质量比独立地选自1: 20、1:40、1:60、1:80、1:100、1:120、1:140、1:160、1:180、 1:200中的任意值或任意两者之间的范围值。
[0054]
可选地,所述二硝基化合物与所述还原剂的摩尔比独立地选自1:1、 1:1.2、1:1.3、1:1.4、1:1.5中的任意值或任意两者之间的范围值。
[0055]
可选地,步骤s2中,所述二硝基化合物与所述第二催化剂的质量比为1:(0.02~0.4)。
[0056]
可选地,步骤s2中,所述二硝基化合物与所述第二催化剂的质量比独立地选自1:0.02、1:0.04、1:0.06、1:0.08、1:0.10、1:0.20、1: 0.30、1:0.40中的任意值或任意两者之间的范围值。
[0057]
可选地,步骤s2中,所述第二溶剂选自甲醇、乙醇、丙酮中的至少一种;
[0058]
所述第二催化剂为pd/c;
[0059]
所述还原剂为水合肼。
[0060]
可选地,步骤s2中,反应 在回流下进行;
[0061]
反应 的条件如下:
[0062]
温度为;60~100;
[0063]
时间为2h~60h。
[0064]
可选地,步骤s2中,反应 在非活性气氛下进行;
[0065]
所述非活性气氛选自氮气、氩气中的至少一种。
[0066]
反应 后还需进行后处理,包括减压蒸馏、洗涤、干燥、分离及纯化。
[0067]
步骤s1中在冰水浴条件下分别将溶有式(4)所示羟基化合物溶液与溶有式(5)所示化合物溶液滴入体系中,滴加完毕后除去冰浴,室温搅拌,室温搅拌反应时间为2~24小时,优选为4~10小时。
[0068]
优选地,步骤s1的纯化处理包括洗涤、过滤、干燥。优选地,洗涤液为质量比为1:1的醋酸乙酯、正己烷、石油醚或乙醚和水。
[0069]
作为优选的技术方案,所述二胺的制备方法包括如下步骤:
[0070]
步骤一:在氮气保护下,向装有搅拌的三口烧瓶中加入第一催化剂,用第一溶剂将其溶解后,置于冰水浴中,将溶有式(4)所示羟基化合物的溶液滴加入体系中,再将溶有式(5)所示卤代化合物溶液滴入体系,滴加完成后除去冰水浴,自然升温至室温,继续搅拌4~
10小时后,加入洗涤液终止反应,经洗涤液进一步清洗、过滤并干燥,得到式(6)所示含有苯并环丁烯的二硝基化合物。
[0071]
步骤二:在氮气保护下,将式(6)所示二硝基化合物加入装有搅拌的三口烧瓶中,加入第二溶剂、催化剂,搅拌均匀后加热至90,滴加还原剂,回流2~60小时后,停止加热,自然冷却至室温,过滤、重结晶,再过滤,重结晶、过滤,得到式(1)所示二胺。
[0072]
根据本技术的第四个方面,提供了一种聚酰亚胺前体的制备方法。
[0073]
上述所述聚酰亚胺前体的制备方法,包括以下步骤:
[0074]
将含有二胺化合物、四羧酸二酐、有机溶剂的混合物,反应 得到所述聚酰亚胺前体;
[0075]
所述二胺化合物选自权利要求1所述的二胺化合物。
[0076]
可选地,混合物中还包括其他二胺化合物;
[0077]
其他二胺化合物选自4,4
’‑
二氨基二苯甲烷、4,4
’‑
二氨基二苯乙烷、 4,4
’‑
二氨基二苯酮、3,4
’‑
二氨基二苯酮、3,3
’‑
二氨基二苯酮、4,4
’‑ꢀ
二氨基-2,2
’‑
二甲基联苯、4,4
’‑
二氨基二苯基砜、4,4
’‑
二氨基二苯醚、 4,4
’‑
二氨基-3,3
’‑
二甲基联苯、4,4
’‑
二氨基二苯硫醚、2,2
’‑
二[4-(4
‑ꢀ
氨基苯氧基)苯基]六氟丙烷、2,2
’‑
二[4-(4-氨基苯氧基)苯基]丙烷、2,2
’‑ꢀ
二(4-氨基苯基)丙烷、2,2
’‑
二(4-氨基苯基)六氟丙烷、对苯二胺、间苯二胺、丁二胺、戊二胺、己二胺、庚二胺、辛二胺中的至少一种。
[0078]
可选地,所述二胺化合物占总的二胺的1%~70%;
[0079]
总的二胺指所述二胺化合物和其他二胺化合物的总和。
[0080]
可选地,所述有机溶剂选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、 n-甲基吡咯烷酮、n-甲基己内酰胺、二甲基亚砜、γ-丁内酯、甲乙酮、环己酮、环戊酮、4-羟基-4-甲基-2-戊酮中的至少一种。
[0081]
聚酰亚胺前体聚酰胺酸可通过含有本发明二胺与四羧酸二酐在有机溶剂中进行反应得到。
[0082]
上述有机溶剂只要能溶解所生成的聚酰胺酸即可,具体列举为n,n
‑ꢀ
二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、n-甲基己内酰胺、二甲基亚砜、γ-丁内酯等。另外,聚酰亚胺前体的溶解性高时,可以使用甲乙酮、环己酮、环戊酮、4-羟基-4-甲基-2-戊酮。
[0083]
上述有机溶剂可单独使用也可混合使用。由于有机溶剂中的水分会抑制聚合反应,因此,有机溶剂可进行脱水干燥后使用。
[0084]
作为二胺与四羧酸二酐混合在有机溶剂中进行反应的方法,可列举为:搅拌使二胺分散或溶解于有机溶剂而得到的溶液,直接添加羧酸二酐或者使四羧酸二酐分散或溶解于有机溶剂后添加的方法;向四羧酸二酐分散或溶解于有机溶剂而得到的溶液中添加二胺的方法;将羧酸二酐和二胺交替或同时添加至有机溶剂的方法等,可以是上述方法中的任意一种。
[0085]
可选地,所述四羧酸二酐与所述二胺化合物的摩尔比为1:0.8~1.2。
[0086]
可选地,反应 的条件如下:
[0087]
温度为;-20~150;
[0088]
时间为1h~72h。
[0089]
可选地,反应 的条件如下:
[0090]
温度为;0~80;
[0091]
时间为2h~48h。
[0092]
反应浓度优选为1~50%,更优选为5~20%。反应浓度过低时,难以得到分子量高的聚合物,反应浓度过高时,反应液的粘度过大导致难以搅拌均匀。优选地,可以在反应初期以高浓度进行,在反应后期追加有机溶剂。
[0093]
通常情况下,该摩尔比越接近1:1,则生成的聚酰胺酸分子量越大。
[0094]
为了获得较好的涂敷性能,在反应后期追加的有机溶剂可以是n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和γ
‑ꢀ
丁内酯等良溶剂至少一种,还可以包含乙基溶纤剂、丁基溶纤剂、二甘醇-乙醚、二甘醇-丁醚、二甘醇-乙醚乙酸酯、乙二醇、1-甲氧基-2-丙醇、 1-乙氧基-2-丙醇、1-丁氧基-2-丙醇、1-苯氧基-2-丙醇、丙二醇乙酸酯、丙二醇二乙酸酯、二丙二醇、乳酸乙酯中的至少一种;
[0095]
根据本技术的第五个方面,提供了一种液晶取向剂。
[0096]
一种液晶取向剂,所述液晶取向剂包括聚合物a和溶剂b;
[0097]
所述聚合物a选自聚酰亚胺前体、聚酰亚胺中的至少一种;
[0098]
所述聚酰亚胺由所述聚酰亚胺前体亚胺化得到;
[0099]
所述聚酰亚胺前体选自上述所述的聚酰亚胺前体和/或上述所述的制备方法得到的聚酰亚胺前体。
[0100]
可选地,所述聚合物a的使用量为100重量份;
[0101]
所述溶剂b的使用量为500~5000重量份。
[0102]
所述溶剂b选自n-甲基吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和γ-丁内酯、乙基溶纤剂、丁基溶纤剂、二甘醇-乙醚、二甘醇-丁醚、二甘醇-乙醚乙酸酯、乙二醇、1-甲氧基-2-丙醇、1-乙氧基-2-丙醇、1-丁氧基-2-丙醇、1-苯氧基-2-丙醇、丙二醇乙酸酯、丙二醇二乙酸酯、二丙二醇、乳酸乙酯中的至少一种。
[0103]
从聚合物分子量的角度考虑,所述聚合物由二酐组分和二胺组分反应而成,其中二酐组分与二胺组分的摩尔比为100:(20-200)。
[0104]
为了保证取向剂的预倾角在合适的范围内,优选的,上述二胺2,2
’‑ꢀ
二[4-(4-氨基苯氧基)苯基]六氟丙烷、2,2
’‑
二[4-(4-氨基苯氧基)苯基]丙烷、2,2
’‑
二(4-氨基苯基)丙烷、2,2
’‑
二(4-氨基苯基)六氟丙烷与上述二酐4,4'
‑ꢀ
(4,4'-异丙基二苯氧基)二酞酸酐、4,4
′‑
(六氟异丙烯)二酞酸酐、4,4'-异亚丙基-双(邻苯二甲酸酐)、4,4'-(4,4'-异丙基二苯氧基)二酞酸酐的总摩尔量占聚酰亚胺前体或聚酰亚胺中二胺与二酐总摩尔量的30%~70%。
[0105]
进一步的,4,4'-(4,4'-异丙基二苯氧基)二酞酸酐、4,4
′‑
(六氟异丙烯) 二酞酸酐、4,4'-异亚丙基-双(邻苯二甲酸酐)、4,4'-(4,4'-异丙基二苯氧基) 二酞酸酐占聚酰亚胺前体或聚酰亚胺中二酐总摩尔量的0~50%。
[0106]
更进一步的,2,2
’‑
二[4-(4-氨基苯氧基)苯基]六氟丙烷、2,2
’‑
二(4-氨基苯基)六氟丙烷与上述二酐4,4
′‑
(六氟异丙烯)二酞酸酐的总摩尔量占聚酰亚胺前体或聚酰亚胺中二胺与二酐总摩尔量的30%~70%。
[0107]
根据本技术的第六个方面,提供了一种液晶取向剂的应用。
[0108]
上述所述的液晶取向剂在液晶取向膜、液晶显示元件中的应用。
[0109]
本发明还提供了一种液晶取向膜,其是由上述液晶取向剂所制。
[0110]
优选地,该液晶取向膜的形成方式包含的步骤为:将上述液晶取向剂利用滚涂法、旋涂法、印刷法、喷墨法(ink-jet)等方法,涂布在基材的表面上,形成预涂层,接着将该预涂层经过预固化、主固化及摩擦配向处理而值得。
[0111]
该预固化的目的在于使该预涂层中的有机溶剂挥发。该预固化的操作温度范围为30~120,优选为40~110,更优选为50~100。
[0112]
该主固化的目的在于使该预涂层中的聚合物再进一步进行脱水闭环 (酰亚胺化)反应。该主固化的操作温度范围为150~300,优选为 180~280,更优选为200~250。
[0113]
该摩擦配向处理并无特别的限制,可采用尼龙、人造丝、棉类等纤维所制成的布料缠绕在滚筒上,以一定方向摩擦进行配向。上述摩擦配向处理为本领域的一般技术人员所周知,因此不再多加赘述。
[0114]
本发明还提供了一种液晶显示器,该液晶显示器件包括上述液晶取向膜。该液晶显示器件可以是tn-lcd、stn-lcd、ips-lcd、ffs
‑ꢀ
lcd。该液晶显示器件具有vhr高、蓄积电荷低、预倾角稳定性好的优点,且功耗低、信赖性好,对比度高,显示性能优异,有效改善液晶显示器的闪烁、低压显示不匀、封口污染等显示问题。
[0115]
聚酰胺酸酯的制备方法
[0116]
1)由聚酰胺酸制备
[0117]
聚酰胺酸脂可通过上述聚酰胺酸制备酯化反应的制备方法:
[0118]
具体为,通过使聚酰胺酸与酯化剂在有机溶剂的存在以-20~ 150、优选为0~50反应0.5~24小时,优选为1~4小时来制备。
[0119]
作为酯化剂,优选可通过精制而容易除去的酯化剂,列举为n,n-二甲基甲酰胺二甲基乙缩醛、n,n-二甲基甲酰胺二乙基乙缩醛、n,n-二甲基甲酰胺二丙基乙缩醛、n,n-二甲基甲酰胺二新戊基丁基乙缩醛、n,n
‑ꢀ
二甲基甲酰胺二叔丁基乙缩醛、1-甲基-3-甲苯基三氮烯、1-乙基-3-对甲苯基三氮烯、1-丙基-3-对甲苯基三氮烯、4-(4,6-二甲氧基-1,3,5-三嗪-2
‑ꢀ
基)-4-甲基吗啉氯等,酯化剂的添加量为聚酰胺酸重复单元摩尔比为2-6: 1。
[0120]
有机溶剂只要能溶解所生成的聚酰胺酸即可,具体列举为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、n-甲基己内酰胺、二甲基亚砜、γ-丁内酯等。另外,聚酰亚胺前体的溶解性高时,可以使用甲乙酮、环己酮、环戊酮、4-羟基-4-甲基-2-戊酮。
[0121]
上述有机溶剂可单独使用也可混合使用。由于有机溶剂中的水分会抑制聚合反应,因此,有机溶剂可进行脱水干燥后使用。从聚合物溶解性的角度出发,上述反应中使用的有机溶剂优选为n,n-二甲基甲酰胺、 n-甲基吡咯烷酮或γ-丁内酯,可以使用一种或混合2种以上。从聚合物不易析出且高分子量的观点出发,反应浓度优选为1~30wt%、更优选为 5~20wt%。
[0122]
2)通过四羧酸二酯二氯化合物与二胺进行反应的制备方法:
[0123]
聚酰胺酸脂可以通过四羧酸二酯二氯化合物与含有本发明二胺的二胺进行制备。
[0124]
具体为,通过四羧酸二酯氯化物与二胺在碱和有机溶剂的存在下以
‑ꢀ
20~150、优选0~50反应30~24小时、优选为1~4小时来制备。
[0125]
作为碱,可以使用吡啶、三乙胺、4-二甲氨基吡啶等,为了使反应温和地进行,优选
为吡啶。从容易除去且容易获得高分子量的观点出发,碱的添加量相当于四羧酸二酯二氯化物优选2~4倍摩尔、更优选为2~ 3倍摩尔。
[0126]
从单体和聚合物的溶解性的观点出发,用于上述反应的溶剂优选为 n-甲基吡咯烷酮或γ-丁内酯,可以使用上述一种或混合2种以上溶剂。从聚合物不易析出且高分子量的观点出发,反应浓度优选为1~30wt%、更优选为5~20wt%。
[0127]
为了防止四羧酸二酯二氯化物的水解,用于制造聚酰胺酸酯的溶剂优选为脱水溶剂,在氮气保护下进行,防止大气混入。
[0128]
3)由四羧酸二酯和二胺来制备的方法
[0129]
聚酰胺酸脂可以通过四羧酸二酯与含有本发明的二胺成分进行缩聚来制备。
[0130]
具体的,可以通过使用四羧酸二酯与二胺在缩合剂、碱和有机溶剂的存在下以0~150、优选0~100反应30分钟到24小时、优选为3 ~15小时来制备。
[0131]
作为缩合剂,可以使用亚磷酸三苯酯、二环己基碳二亚胺、1-乙基
‑ꢀ
3-(3-二甲基丙基)碳二亚胺盐酸盐、n,n
’‑
羰基二咪唑、二甲基-1,3,5-三嗪基甲基吗啉、0-(苯并三唑-1-基)-n,n,n’,n
’‑
四甲基脲四氟硼酸盐、0-(苯并三唑-1-基)n,n,n’,n
’‑
四甲基脲六氟磷酸盐、(2,3-二氢-2-硫杂-3-苯并噁唑基)磷酸二苯基酯等。缩合剂的添加量相对于四羧酸二酯优选为2~3倍摩尔量。
[0132]
作为碱,可以使用吡啶、三乙胺等叔胺。从易除去和易得到高分子量的观点出发,碱的添加量相对于二胺成分优选为2~4倍摩尔量。
[0133]
另外,上述反应中,通过添加路易斯酸作为添加剂,反应会有效地进行。作为路易斯酸,优选为氯化锂、溴化锂等卤代锂。路易斯酸的添加量相对于二胺成分优选为0~1.0倍摩尔量。
[0134]
上述3中聚酰胺酸脂的制备方法中,为了更容易得到高分子量的聚酰胺酸脂,优选上述1)或上述2)所述制备方法。
[0135]
将上述方法得到的聚酰胺酸脂溶液通过一边搅拌一边注入不良溶剂中,使聚合物析出。进行数次析出并用不良溶剂清洗后,进行常温干燥或加热干燥,能够得到经精制的聚酰胺酸脂粉末。不良溶剂没有特别限定,可列举出水、甲醇、乙醇、乙烷、丁基溶纤剂、丙酮、甲苯等。
[0136]
聚酰亚胺的制备方法
[0137]
本发明中的聚酰亚胺可通过前述聚酰亚胺前体酰亚胺化来制备。
[0138]
本发明的聚酰亚胺酰亚胺化率可以不是100%,可根据使用要求进行调整。
[0139]
所为使聚酰亚胺前体酰亚胺化的方法,可列举出不适用催化剂而加热聚酰亚胺前体的热亚胺化方法、使用催化剂的催化酰亚胺化。
[0140]
使用聚酰亚胺前体进行热酰亚胺化时,优选的,将聚酰亚胺前体的溶液加热至100~400、优选为120~200,一边将酰亚胺化反应产生的水/醇除去体系外,一边进行热酰亚胺化。
[0141]
聚酰亚胺前体的催化酰亚胺化可通过向聚酰胺酸的溶液中添加碱性催化剂和酸酐,以-20~250、优选0~180进行搅拌来进行。碱性催化剂的量为酰胺酸基的0.5~30摩尔量、优选为2~20倍摩尔量,酸酐的量为酰胺酸基的1~50倍摩尔量、优选为3~30倍摩尔量。
[0142]
作为碱性催化剂,可列举出吡啶、三乙胺、三甲胺、三丁胺、三辛胺等,其中,由于具
有适合推进反应的碱性,因此优选吡啶。
[0143]
作为酸酐,可列举出醋酸酐、偏苯三酸酐、均苯四酸酐等,其中,使用醋酸酐时,反应结束后容易进行精制,因此优选为醋酸酐。
[0144]
进一步的,催化酰亚胺化的酰亚胺化率可通过调整催化剂量和反应温度、反应时间来控制。
[0145]
从聚酰亚胺的反应溶液回收聚合物成分时,将反应溶液投入不良溶剂而使其沉淀即可。该不良溶剂可列举出甲醇、丙酮、己烷、丁基溶纤剂、庚烷、甲乙酮、甲基异丁基酮、乙醇、甲苯、苯、水等。投入不良溶剂使其沉淀而得到的聚合物优选为过滤回收以后常压或减压进行常温干燥或加热来干燥。
[0146]
液晶取向剂的制备方法
[0147]
该液晶取向剂的制备方法并无特别的限制,可采用一般的混合方法,如先将聚酰胺酸、聚酰胺酸脂、聚酰亚胺混合均匀,形成聚合物a,接着,再将该聚合物a在温度为0至200的条件下加入溶剂b,一搅拌装置持续搅拌至溶解即可。优选地,在20至60的条件下,将聚合物 a添加至溶剂b中。
[0148]
本技术能产生的有益效果包括:
[0149]
1)本技术所提供的一种二胺化合物。该二胺化合物为一种新型的含苯并环丁烯的二胺,由于苯并环丁烯在200以上加热,环丁烯结构发生开环反应,形成八元环结构,阻断苯环间电荷传输,提高电绝缘性,含有该二胺单体的聚酰亚胺电阻率高,介电常数低。同时苯并环丁烯间的交联作用,进一步使聚酰亚胺薄膜的力学性能,耐摩擦性能明显提高。
[0150]
2)本技术所提供的一种液晶取向剂,通过液晶取向剂中关键成分的选择,即通过本发明的液晶取向剂,形成液晶取向膜,机械性能优异,电绝缘性佳,在实施摩擦处理时不易因摩擦处理而产生摩擦斜纹,耐摩擦性好、通过本发明液晶取向剂所制液晶显示器件的vhr高、rdc低、预倾角稳定性高、功耗低、可靠性高、改善封口污染、有效提高lcd的良品率。
附图说明
[0151]
图1为本技术液晶盒封口污染测试中,封口无污染的实物图。
[0152]
图2为本技术液晶盒封口污染测试中,封口轻微污染的实物图。
[0153]
图3为本技术液晶盒封口污染测试中,封口严重污染的实物图。
具体实施方式
[0154]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0155]
如无特别说明,本技术的实施例中的原料和催化剂均通过商业途径购买。
[0156]
下述实施例使用的成分简称如下:
[0157]
cbda:1,2,3,4-环丁烷四羧酸二酐
[0158]
tca:3-羧甲基-1,2,4-环戊烷三甲酸1,4:2,3-双酐
[0159]
hfda:4,4
′‑
(六氟异丙烯)二酞酸酐
[0160]
bpada:4,4'-(4,4'-异丙基二苯氧基)二酞酸酐
[0161]
ddm:4,4
’‑
二氨基二苯基甲烷
[0162]
bapp:2,2
’‑
双[4-(4-氨基苯氧基)苯基]-丙烷
[0163]
hfbapp:2,2
’‑
双[4-(4-氨基苯氧基)苯基]-1,1,1,3,3,3-六氟丙烷
[0164]
sida:1,3-双(3-氨基丙基)四甲基二硅氧烷
[0165]
b-1:式(b-1)所示结构
[0166]
b-2:式(b-2)所示结构
[0167]
b-3:式(b-3)所示结构
[0168]
b-4:式(b-4)所示结构
[0169]
b-5:式(b-5)所示结构
[0170]
b-6:式(b-6)所示结构
[0171]
nmp:n-甲基吡咯烷酮
[0172]
bc:丁基溶纤剂
[0173][0174]
合成例1
[0175]
b-1的合成
[0176][0177]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)
0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有
①
(0.105mol,19.54g)的50克四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500克醋酸乙酯和500克超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500克醋酸乙酯和500克超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
②
24.30g,收率84.9%。
[0178][0179]
在氮气保护下,将二硝基化合物
②
24.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,1.2g pb/c催化剂,搅拌均匀后升温至90,30 min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-1 15.70g,82.7%。
[0180]
b-1核磁信息如下:
[0181]1hnmr(dmso):δ:2.88(m,4h),5.27(s,4h),6.03(s,1h),6.19(d,1h), 6.70(d,1h),6.93(s,1h),7.01(d,1h),7.23(d,1h)。
[0182]
合成例2
[0183]
b-2的合成
[0184][0185]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有
③
(0.105mol,21.00g)的50g四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500g醋酸乙酯和500g超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500g醋酸乙酯和500g超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
④
25.70g,收率85.6%。
[0186][0187]
在氮气保护下,将二硝基化合物
④
25.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,1.00g pb/c催化剂,搅拌均匀后升温至90, 30min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-2 17.20g,86.0%。
[0188]
b-2核磁信息如下:
[0189]1hnmr(dmso):δ:2.25(s,3h),2.88(m,4h),5.32(s,2h),6.60(d,1h), 6.74(d,1h),6.93(s,1h),7.01(d,1h),7.23(d,1h),7.51(s,2h)。
[0190]
合成例3
[0191]
b-3的合成
[0192][0193]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有
⑤
(0.0525mol,10.71g)的50g四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500g醋酸乙酯和500g超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500g醋酸乙酯和500g超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
⑥
17.70g,收率87.5%。
[0194][0195]
在氮气保护下,将二硝基化合物
⑥
17.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,0.70g pb/c催化剂,搅拌均匀后升温至90, 30min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-3 12.31g,85.0%。
[0196]
b-3核磁信息如下:
[0197]1hnmr(dmso):δ:2.88(m,8h),5.27(s,4h),5.99(s,1h),6.35(s,1h), 6.93(s,2h),7.01(d,2h),7.23(d,2h)。
[0198]
合成例4
[0199]
b-4的合成
[0200][0201]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有
⑦
(0.105mol,24.80g)的50g四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500g醋酸乙酯和500g超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500g醋酸乙酯和500g超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
⑧
29.11g,收率86.6%。
[0202][0203]
在氮气保护下,将二硝基化合物
⑧
28.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,1.20g pb/c催化剂,搅拌均匀后升温至90, 30min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-4 20.15g,87.6%。
[0204]
b-4核磁信息如下:
[0205]1hnmr(dmso):δ:2.88(m,4h),4.62(s,2h),5.59(s,2h),6.01(d,1h), 6.75(d,1h),6.93(s,1h),7.01(m,2h),7.13(s,1h),7.23(d,1h),7.44(d,1h)。
[0206]
合成例5
[0207]
b-5的合成
[0208][0209]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有
⑨
(0.0525mol,14.71g)的50g四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500g醋酸乙酯和500g 超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500g醋酸乙酯和500g超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
⑩
20.32g,收率84.6%。
[0210][0211]
在氮气保护下,将二硝基化合物
⑨
20.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,0.90g pb/c催化剂,搅拌均匀后升温至90, 30min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-5 14.96g,85.5%。
[0212]
b-5核磁信息如下:
[0213]1hnmr(dmso):δ:2.88(m,8h),5.49(s,4h),6.69(m,4h),6.93(s,2h), 7.01(d,2h),7.23(d,2h),7.58(d,2h)。
[0214]
合成例6
[0215]
b-6的合成
[0216][0217]
在氮气保护下,向装有搅拌的三口烧瓶中加入氢化钠(60wt%的矿物油分散液)0.12mol 4.8g加入四氢呋喃50g,置于冰浴中进行搅拌,将溶有4-羟基-苯环丁烯(0.1mol,12.02g)的150g四氢呋喃溶液缓慢滴入三口烧瓶中,再将溶有(0.0525mol,15.23g)的50g四氢呋喃溶液滴加到三口烧瓶内,在室温下搅拌6小时后,添加500g醋酸乙酯和500g 超纯水,在室温下对析出的晶体进行减压过滤,将得到的固体用500g醋酸乙酯和500g超纯水进一步进行清洗,减压过滤并干燥,得到二硝基化合物
⑩
21.57g,收率86%。
[0218][0219]
在氮气保护下,将二硝基化合物
⑨
20.00g加入装有搅拌的三口烧瓶中,加入乙醇1000ml,0.90g pb/c催化剂,搅拌均匀后升温至90, 30min内滴加完水合肼50ml,回流48h后自然冷却至室温,过滤,重结晶后再过滤,干燥得b-6 15.26g,86.8%。
[0220]
b-6核磁信息如下:
[0221]1hnmr(dmso):δ:2.88(m,8h),5.22(s,4h),6.45(m,4h),6.81(d,2h), 6.93(s,2h),7.01(d,2h),7.23(d,2h)。
[0222]
液晶取向剂的制备
[0223]
在实施例或对比例中,聚酰胺酸溶液的粘度使用椎板粘度计 (brookfield dv2t rv),在温度25的条件下进行测定。
[0224]
实施例1
[0225]
将181.46g nmp投入到带有氮气保护和搅拌的三口烧瓶中,开启搅拌,依次加入二胺单体b-1 0.57g(2.5mmol)、bapp 10.06g(24.5mmol)、 hfbapp 10.06g(22.5mmol)、sida 0.12g(0.5mmol),混合均匀后,加入 cbda 9.81g(50.0mmol),常温下反应24小时后加入nmp和bc,配成 5wt%,nmp为55wt%、bc为40wt%的溶液,得到液晶取向剂a-1。实施例2~14和对比例1~8中聚合物所用单体的种类及用量有所改变,具体见表1、表2所示,其他同实施例1。
[0226]
表1
[0227]
[0228][0229]
一种液晶显示元件
[0230]
将液晶取向剂用ptfe制的膜过滤器过滤,再用旋涂法将其涂布在带 ito电极和
6um spacer的玻璃基板上(玻璃基板购买自fpd solutions),形成预涂层。经过预固化(热台、80,5分钟)、主固化(循环烘箱, 230,60分钟)、将得到膜厚为80nm的液晶取向膜。再将薄膜通过辊轮直径120mm的人造丝布摩擦装置,以固定转速600rpm、基板进行速度20mm/s、压入量0.3mm的条件对膜表面进行摩擦取向处理,得到包含液晶取向膜的基板。印刷密封框胶,使得液晶配向膜相对于摩擦方向 180度的帖向另一片基板灌入液晶,制成反平行液晶显示元件。
[0231]
(1)耐摩擦性
[0232]
将实施例和比较例中所得的液晶显示元件的上下基板贴上偏振轴方向正交的偏光片,然后将液晶显示元件放置于背光板上面,用5倍的放大镜观察阈值附近电压(液晶显示元件相对透过率为10%附近的电压) 点亮的液晶显示器件的摩擦条纹。摩擦条纹越轻微说明取向膜的耐摩擦性能越好。
[0233]
耐摩擦性能的评价结果如下:
[0234]
放大镜观测无摩擦条纹,耐摩擦性能良好,判定为优;
[0235]
放大镜观测有轻微摩擦条纹,肉眼观测不到摩擦条纹,耐摩擦性能一般,判定为良;
[0236]
肉眼观测到摩擦条纹,耐摩擦性能差,判定为差;
[0237]
(2)介电常数测试
[0238]
利用匀胶机将实施例1~14和对比例1~8的液晶取向剂分别均匀涂敷在硅晶片上,经过预固化(热台、80,5分钟)、主固化(循环烘箱, 230,60分钟)、将得到膜厚为10~20um的薄膜。将带膜的硅晶片浸泡在47%的氢氟酸溶液中5~10分钟,然后用水冲洗,得到聚酰亚胺薄膜。将薄膜切割成长80mm、宽60mm的样条,采用ne5224b型号电路网分析仪,利用共振器扰动法算出1mhz下的介电常数。
[0239]
(3)预倾角及其稳定性
[0240]
通过结晶旋转法来测量液晶显示器件中心点在23下的预倾角,再升温至60时,经过240小时后得预倾角(p
ht
),且与23下所测得的预倾角(p
rt
)进行比较,通过公式s(%)=(p
ht-p
rt
)/p
rt
×
100%计算预倾角稳定性(s),并根据以下基准进行评价:
[0241]
s小于0.5%,判定为优;0.5%≤s<1.5%,判定为良;1.5%≤s,判定为差。
[0242]
(4)电压保持特性
[0243]
对于所述制造的扭曲向列型液晶显示器件,施加5v的电压60us,测定16.67ms后的电压,算出从初始值开始变化作为电压保持率。测定时,将液晶盒的温度设定为23、90,分别在这2种温度下进行测定。
[0244]
(5)电荷积累特性(施加直流电压后的残留电压)
[0245]
在23条件下,对测定了电压保持率特性的扭转向列型液晶器件施加叠加3v直流电压的
±
3v/30hz的矩形波60分钟,用光学闪烁消除法测定3v直流电压刚切断后的液晶器件内所残留的残余电压。
[0246]
(6)功耗测试
[0247]
将液晶显示元件在25下,以2.3v,32hz的方形波对其进行功耗测试。
[0248]
(7)液晶盒封口污染测试
[0249]
对于所述制造的扭曲向列型液晶显示器件施加2.5v直流电压60s,切断电源,观察
液晶显示盒封口污染情况,封口无污染(如图1),判定为优;封口轻微污染(如图2),判定为良;封口严重污染(如图3),判定为差。
[0250]
(1)~(6)测试结果见表2
[0251]
表2
[0252][0253][0254]
从上述表2中的实施例1~4可以看出,当本发明二胺含量大于等于 15%,液晶取
向膜具有优异的耐摩擦性和较低的介电常数,液晶显示器件预倾角稳定性优、且具有较高的vhr和较低的rdc,无明显封口污染情况,且随着b-1含量的增大电学性能更加优异。说明含有本发明b-1二胺的液晶取向剂的加入可明显提高了液晶显示器件的良品率。
[0255]
通过表2中实施例2、7~14与对比例1~8可以看出,含有本发明二胺的液晶取向膜耐摩擦性能优异,且具有较低的介电常数,可明显提升液晶显示器件的电学性能,降低液晶显示器的功耗,同时液晶显示器件的预倾角稳定性优。在相同二胺含量摩尔比下,二胺b-3、bapp、 hfbapp、sida与tca制备的液晶取向剂在相同本发明二胺含量的情况下具有较佳的电学性能。
[0256]
因此,本发明液晶取向剂可明显提高液晶取向膜的耐摩擦性、且介电常数低、预倾角稳定性好、vhr高、rdc低、功耗低,可明显改善液晶显示器件的封口污染、液晶冲刷纹、摩擦痕迹、画面闪烁等问题,提高液晶显示器件的可靠性和良品率。
[0257]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1