一种吡唑衍生物及其制备方法与应用
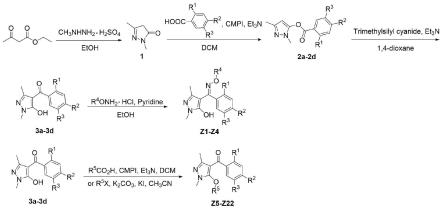
1.本发明属于化合物合成技术领域,具体涉及一种吡唑衍生物及其制备方法与应用。
背景技术:
2.杂草是农田生态系统的重要组成之一,其与病害、虫害并列,对农业生产造成严重的危害。杂草与作物幼苗争夺阳光、空间、水和营养,导致作物减产及农产品品质降低。除草剂的使用可以有效地提高作物产量,这为除草剂的发展提供了机会,不断研发新型高效对环境友好除草剂是当前农业生产的迫切需求。
3.近年来,具有吡唑片段的小分子被广泛应用于医药、农药、材料等领域,其中在农药领域,吡唑类化合物表现出多样的生物活性,例如抗细菌、抗真菌、除草等。同时,由于其高效、低毒以及吡唑环上取代基多方位变换而在农药中扮演着十分重要的角色。大量文献的持续报道且不断有新的吡唑类农药商品化。目前,吡唑类化合物已成为新农药创制研究中令人关注的焦点之一。吡草酮、磺酰草吡唑、吡唑特、苯唑草酮、苄草唑、异丙吡草酯、吡草醚、吡嘧磺隆、双唑草腈等商品除草剂中皆含有吡唑结构,其中,大部分为芳酰基吡唑结构,属于对羟基苯丙酮酸双加氧酶(hppd)抑制剂,该类抑制剂具有除草谱广、作物选择性好和能有效防治对草甘膦等除草剂产生抗性的杂草等优势。由青岛清原农冠公司研发的环吡氟草酮和双唑草酮,首次将hppd抑制剂吡唑类化合物引入到小麦田抗性禾本科和阔叶杂草的防治上,完美解决了乙酰乳酸合成酶和乙酰辅酶a羧化酶抑制剂的抗性和多抗性问题。因此,基于芳酰基吡唑单元设计合成除草药物小分子是十分符合当前新农药创制的趋势。
4.申请号为200980149775.2和200780043787.8公开了一种可用于制作除草药物的苯甲酰吡唑化合物或其盐,该化合物具有更宽除草谱且具有高度有效和持续长时间除草活性的除草组合物,但在安全性和生物降解性方面有待提升。
技术实现要素:
5.本发明针对现有技术的不足,提出了一种吡唑衍生物及其制备方法与应用。
6.具体是通过以下技术方案来实现的:
7.一种吡唑衍生物为含肟醚基的吡唑衍生物或含苯甲酰基的吡唑衍生物;
8.一种含肟醚基的吡唑衍生物,是将吡唑苯甲酰上的羰基与o-取代基羟胺反应生成肟醚,其结构通式如式(i):
9.10.式中:r1为氯原子;
11.r2为氯原子、甲砜基;
12.r3为氢原子;
13.r4为苄基、烯丙基;
14.另一种含苯甲酰基的吡唑衍生物是吡唑上的羟基被不同基团取代,其结构通式如式(ii):
[0015][0016]
式中:r1为氯原子、硝基、氟原子;
[0017]
r2为氯原子、氢原子、甲砜基;
[0018]
r3为氢原子、氯原子;
[0019]
r5为1-甲基-3-三氟甲基吡唑甲酰基、4-硝基苯磺酰基、乙基异丙基碳酸酯基、环丙基异噁唑甲酰基、1,3-二甲基吡唑甲酰基。
[0020]
所述吡唑衍生物,包括如下化合物:
[0021]
化合物z1:(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚;
[0022]
化合物z2:(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚;
[0023]
化合物z3:(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚;
[0024]
化合物z4:(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚;
[0025]
化合物z5:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯;
[0026]
化合物z6:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯;
[0027]
化合物z7:1-((4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯;
[0028]
化合物z8:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯;
[0029]
化合物z9:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯;
[0030]
化合物z
10
:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯;
[0031]
化合物z
11
:1-((4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯;
[0032]
化合物z
12
:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯;
[0033]
化合物z
13
:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯;
[0034]
化合物z
14
:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯;
[0035]
化合物z
15
:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯;
[0036]
化合物z
16
:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯;
[0037]
化合物z
17
:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯;
[0038]
化合物z
18
:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯;
[0039]
化合物z
19
:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯;
[0040]
化合物z
20
:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯;
[0041]
化合物z
21
:1-((4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯;
[0042]
化合物z
22
:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯。
[0043]
本发明的另一个目的在于提供所述吡唑衍生物的制备方法,以乙酰乙酸乙酯、甲基肼硫酸盐为主要原料,分别经环化、加成、重排、缩合或取代反应合成含肟醚及苯甲酰基的吡唑衍生物;所述吡唑衍生物上羟基取代为1-甲基-3-三氟甲基吡唑甲酰基、4-硝基苯磺酰基、乙基异丙基碳酸酯基、环丙基异噁唑甲酰基、1,3-二甲基吡唑甲酰基中任一种。
[0044]
所述肟醚为o-烯丙基肟醚、o-苄基肟醚中任一种。
[0045]
所述苯甲酰基为2,4-二氯苯甲酰基、2-氟-4-氯苯甲酰基、2-氯-4-甲砜基苯甲酰基、2-硝基-5-氯苯甲酰基中任一种。
[0046]
所述吡唑衍生物的制备方法,包括如下步骤:
[0047]
(1)1,3-二甲基吡唑酮(中间体1)的制备:
[0048]
取甲基肼硫酸盐,室温下滴入乙酰乙酸乙酯和乙醇的混合溶液中,滴毕,80℃加热回流,反应体系经减压浓缩、乙酸乙酯萃取、干燥及柱层析后即得固体1,3-二甲基吡唑酮,即为中间体1;
[0049]
(2)1,3-二甲基-1h-吡唑-5-基多取代苯甲酸酯(中间体2a-2d)的制备:
[0050]
取多取代苯甲酸、2-氯-1-甲基吡啶碘化物(cmpi)、三乙胺与二氯甲烷混匀搅拌一段时间后,室温下加入中间体1和三乙胺,使反应体系在室温下反应8-10小时,反应完全后,经饱和碳酸氢钠溶液洗涤、干燥及重结晶后即得1,3-二甲基-1h-吡唑-5-基多取代苯甲酸酯,即为中间体2a-2d;
[0051]
(3)1,3-二甲基-5-羟基-1h-吡唑-4-基多取代苯甲酰(中间体3a-3d)的制备:
[0052]
取中间体2a-2d与1,4-二氧六环混匀搅拌一段时间后,室温下加入三甲基氰硅烷和三乙胺,使反应体系在室温下反应,反应完全后,经萃取、干燥及柱层析即得1,3-二甲基-5-羟基-1h-吡唑-4-基多取代苯甲酰,即为中间体3a-3d;
[0053]
(4)目标化合物含肟醚基团的吡唑衍生物的制备:
[0054]
取中间体3a-3d与乙醇混匀,后在室温下加入o-取代羟胺盐酸盐与吡啶,将反应体系升温至80℃进行回流反应,反应完全后,体系经减压浓缩及柱层析纯化后得到含肟醚基团的吡唑衍生物。
[0055]
(5)目标化合物含苯甲酰基团的吡唑衍生物的制备:
[0056]
取多种羧基有机物与二氯甲烷混匀,在室温下加入cmpi与三乙胺,搅拌一段时间后再加入中间体3a-3d与三乙胺,反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到部分含苯甲酰基团的吡唑衍生物。
[0057]
(6)目标化合物含苯甲酰基团的吡唑衍生物的制备:
[0058]
取中间体3a-3d与碳酸钾在乙腈溶液中混匀,在室温下加入多种取代卤化物与碘化钾,将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到另一部分含苯甲酰基团的吡唑衍生物。
[0059]
在步骤(1)中,所述甲基肼硫酸盐和乙酰乙酸乙酯的用量按摩尔比计为:甲基肼硫酸盐:乙酰乙酸乙酯=3:1。
[0060]
在步骤(1)中,所述乙醇的用量按每毫摩尔的乙酰乙酸乙酯用量加乙醇1ml进行控制。
[0061]
在步骤(2)中,所述中间体1、多取代苯甲酸、cmpi、三乙胺的用量按摩尔比计为:中间体1:多取代苯甲酸:cmpi:三乙胺=2.5:2:2.5:2。
[0062]
在步骤(2)中,所述二氯甲烷的用量按每毫摩尔的中间体1用量加二氯甲烷1ml进行控制。
[0063]
在步骤(3)中,所述中间体2a-2d、三甲基氰硅烷、三乙胺的用量按摩尔比计为:中间体2a-2d:三甲基氰硅烷:三乙胺=1:0.3:1.5。
[0064]
在步骤(3)中,所述1,4-二氧六环的用量按每毫摩尔的中间体2a-2d的用量加1,4-二氧六环1ml进行控制。
[0065]
在步骤(4)中,所述中间体3a-3d、多种o-取代羟胺盐酸盐、吡啶的用量按摩尔比计为:中间体3a-3d:o-取代羟胺盐酸盐:吡啶=1:3:5。
[0066]
在步骤(4)中,所述乙醇的用量按每毫摩尔的中间体3a-3d的用量加乙醇1ml进行控制。
[0067]
在步骤(5)中,所述中间体3a-3d、多种羧基有机物、cmpi、三乙胺的用量按摩尔比计为:中间体3a-3d:多种羧基有机物:cmpi:三乙胺=2.5:2:2.5:2。
[0068]
在步骤(5)中,所述二氯甲烷的用量按每毫摩尔的中间体3a-3d的用量加二氯甲烷1ml进行控制。
[0069]
在步骤(6)中,所述中间体3a-3d、多种卤化物、碳酸钾、碘化钾的用量按摩尔比计为:中间体3a-3d:多种卤化物:碳酸钾:碘化钾=1:3:1.5:0.1。
[0070]
在步骤(6)中,所述乙腈的用量按每毫摩尔的中间体3a-3d的用量加乙腈1ml进行
控制。
[0071]
所述吡唑衍生物的制备路线如下:
[0072][0073]
本发明的再一个目的在于所述吡唑衍生物在制备除草、杂草生长酶抑制药物中的应用。
[0074]
具体的,所述杂草为马唐、稗草、苘麻、反枝苋、小藜、黑麦草、马齿苋、狗牙根、油菜、婆婆纳、苋菜、龙葵、苦麦菜、蒲公英、鼠尾草、三叶草、繁缕、千金子、牛筋草、狗尾草;所述酶为拟南芥对羟基苯丙酮酸双加氧酶(athppd)。
[0075]
有益效果:
[0076]
本发明基于吡唑结构,引入羟胺、苯甲酰基及不同羟基取代基等具有除草活性药效团,因此创制出理化性质相对稳定、成药性优异的含肟醚或苯甲酰基的吡唑类除草药物小分子;该类衍生物尤其对马唐、稗草、苘麻、千金子及牛筋草的苗后抑制效果显著,对苋菜苗前抑制效果显著,且亦对hppd具有优秀的抑制效力。
[0077]
hppd抑制活性测试结果表明化合物z6、z9、z
16
和z
21
对hppd的抑制活性最好,ic50值分别为0.08μm、0.05μm、0.08μm和0.11μm,优于苯唑草酮(1.33μm)和硝磺草酮(1.76μm)。苗前除草活性测试结果表明化合物z1、z9、z
10
、z
11
、z
18
和z
22
在100μg/ml的浓度下对苋菜根茎的抑制率均为100%,抑制效果均优于苯唑草酮(34.8%和40.2%)和硝磺草酮(14.4%和30.4%)。苗后除草活性测试结果表明大多数吡唑化合物在剂量为150g ai ha时,对苘麻具有良好的除草活性,抑制率均在80%以上。化合物z5、z
15
、z
20
和z
21
对稗草和马唐具有优秀的除草活性,在150g ai ha时抑制率达到100%,杂草完全被漂白,生长受到严重抑制。同时,化合物z5、z
15
、z
20
和z
21
在37.5g ai ha的剂量下对稗草的抑制率为100%,与苯唑草酮(100%)一致,但优于硝磺草酮(80%)。
[0078]
本发明含肟醚或苯甲酰基的吡唑衍生物结构简单,制备工艺简单,生产成本低,产率高,低毒、易降解,具有良好的环境相容性,使用安全性高,制备过程无毒无害。
附图说明
[0079]
图1为吡唑衍生物的制备路线图。
具体实施方式
[0080]
下面对本发明的具体实施方式作进一步详细的说明,但本发明并不局限于这些实施方式,任何在本实施例基本精神上的改进或代替,仍属于本发明权利要求所要求保护的范围。
[0081]
实施例1-22提供的吡唑衍生物的制备路线如下:
[0082][0083]
实施例1:(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚(即化合物z1)的制备方法,包括以下步骤:
[0084]
(1)1,3-二甲基吡唑酮的制备:
[0085]
取甲基肼硫酸盐(33.23g,230.52mmol),室温下滴入乙酰乙酸乙酯(10.00g,76.84mmol)和乙醇(100ml)的混合溶液中,滴毕,80℃加热回流,反应体系经减压浓缩、乙酸乙酯萃取、干燥及柱层析后即得固体1,3-二甲基吡唑酮中间体5.00g,收率58.03%。
[0086]
(2)1,3-二甲基-1h-吡唑-5-基-2,4-二氯苯甲酸酯的制备:
[0087]
取2,4-二氯苯甲酸(2.73g,14.27mmol)、cmpi(4.56g,17.84mmol)、三乙胺(1.44g,14.27mmol)与二氯甲烷(20ml)混匀搅拌一段时间后,室温下加入中间体1(2.00g,17.84mmol)和三乙胺(1.44g,14.27mmol),使反应体系在室温下反应8-10小时,反应完全后,经饱和碳酸氢钠溶液洗涤、干燥及重结晶后即得1,3-二甲基-1h-吡唑-5-基2,4-二氯苯甲酸酯中间体1.60g,收率78.65%。
[0088]
(3)1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰的制备:
[0089]
取1,3-二甲基-1h-吡唑-5-基-2,4-二氯苯甲酸酯中间体(2.00g,7.01mmol)与1,4-二氧六环(10ml)混匀搅拌一段时间后,室温下加入三甲基氰硅烷(0.21g,2.10mmol)和三乙胺(1.06g,10.52mmol),使反应体系在室温下反应,反应完全后,经萃取、干燥及柱层析即得1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰中间体1.20g,收率60.00%。
[0090]
(4)目标化合物(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚的制备:
[0091]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰中间体(0.50g,1.75mmol)与乙醇(10ml)混匀,后在室温下加入o-烯丙基羟胺盐酸盐(0.58g,5.26mmol)与吡啶(0.69g,8.77mmol),将反应体系升温至80℃进行回流反应,反应完全后,体系经减压浓缩及柱层析纯化后得到目标化合物(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰
基-o-烯丙基肟醚0.30g,收率50.29%。
[0092]
实施例2:(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚(即化合物z2)的制备方法,包括以下步骤:
[0093]
步骤(1):参照实施例1的步骤(1);
[0094]
步骤(2):参照实施例1的步骤(2),2,4-二氯苯甲酸替换为2-氯-4-甲砜基苯甲酸;
[0095]
步骤(3):参照实施例1的步骤(3);
[0096]
(4)(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚的制备:
[0097]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氯-4-甲砜基苯甲酰中间体(0.80g,2.43mmol)与乙醇(10ml)混匀,后在室温下加入o-烯丙基羟胺盐酸盐(0.80g,7.30mmol)与吡啶(0.96g,12.17mmol),将反应体系升温至80℃进行回流反应,反应完全后,体系经减压浓缩及柱层析纯化后得到目标化合物(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-烯丙基肟醚0.35g,收率37.69%。
[0098]
实施例3:(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚(即化合物z3)的制备方法,包括以下步骤:
[0099]
步骤(1)-(3):参照实施例1的步骤(1)-(3);
[0100]
(4)(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚的制备:
[0101]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰中间体(0.80g,2.81mmol)与乙醇(10ml)混匀,后在室温下加入o-苄基羟胺盐酸盐(1.34g,8.42mmol)与吡啶(1.11g,14.03mmol),将反应体系升温至80℃进行回流反应,反应完全后,体系经减压浓缩及柱层析纯化后得到目标化合物(2,4-二氯苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚0.50g,收率45.66%。
[0102]
实施例4:(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚(即化合物z4)的制备方法,包括以下步骤:
[0103]
步骤(1)-(3):参照实施例2的步骤(1)-(3);
[0104]
(4)(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚的制备:
[0105]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氯-4-甲砜基苯甲酰中间体(0.80g,2.43mmol)与乙醇(10ml)混匀,后在室温下加入o-苄基羟胺盐酸盐(1.17g,7.30mmol)与吡啶(0.96g,12.17mmol),将反应体系升温至80℃进行回流反应,反应完全后,体系经减压浓缩及柱层析纯化后得到目标化合物(2-氯-4-甲砜基苯基)(1,3-二甲基-5-羟基-1h-吡唑-4-基)甲酰基-o-苄基肟醚0.38g,收率35.99%。
[0106]
实施例5:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯(即化合物z5)的制备方法,包括以下步骤:
[0107]
步骤(1)-(3):参照实施例2的步骤(1)-(3);
[0108]
(4)4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯的制备:
[0109]
取1-甲基-3-三氟甲基吡唑-4-甲酸(0.24g,1.22mmol)与二氯甲烷(10ml)混匀,在
4-羧酸酯(即化合物z9)的制备方法,包括以下步骤:
[0125]
步骤(1)-(3):参照实施例1的步骤(1)-(3);
[0126]
(4)4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯的制备:
[0127]
取环丙基异噁唑甲酸(0.13g,0.84mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.27g,1.05mmol)与三乙胺(0.09g,0.84mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰(0.30g,1.05mmol)与三乙胺(0.09g,0.84mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯0.15g,收率84.81%。
[0128]
实施例10:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯(即化合物z
10
)的制备方法,包括以下步骤:
[0129]
步骤(1):参照实施例1的步骤(1);
[0130]
步骤(2):参照实施例1的步骤(2),2,4-二氯苯甲酸替换为2-氟-4-氯苯甲酸;
[0131]
步骤(3):参照实施例1的步骤(3);
[0132]
(4)4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯的制备:
[0133]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氟-4-氯苯甲酰(0.50g,1.86mmol)与碳酸钾(0.38g,2.79mmol)在乙腈溶液(10ml)中混匀,在室温下加入4-硝基苯磺酰氯(0.62g,2.79mmol)与碘化钾(0.03g,0.19mmol),将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯0.37g,收率43.39%。
[0134]
实施例11:1-((4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯(即化合物z
11
)的制备方法,包括以下步骤:
[0135]
步骤(1)-(3):参照实施例8的步骤(1)-(3);
[0136]
(4)1-((4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯的制备:
[0137]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-硝基-5-氯苯甲酰(0.30g,1.01mmol)与碳酸钾(0.21g,1.52mmol)在乙腈溶液(10ml)中混匀,在室温下加入乙基异丙基碳酰氯(0.46g,3.04mmol)与碘化钾(0.02g,0.10mmol),将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到1-((4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯0.20g,收率47.87%。
[0138]
实施例12:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯(即化合物z
12
)的制备方法,包括以下步骤:
[0139]
步骤(1)-(3):参照实施例8的步骤(1)-(3);
[0140]
(4)4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯的制备:
[0141]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-硝基-5-氯苯甲酰(0.30g,1.01mmol)与碳酸钾(0.21g,1.52mmol)在乙腈溶液(10ml)中混匀,在室温下加入4-硝基苯磺酰氯
(0.45g,2.03mmol)与碘化钾(0.02g,0.10mmol),将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯0.25g,收率51.24%。
[0142]
实施例13:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯(即化合物z
13
)的制备方法,包括以下步骤:
[0143]
步骤(1)-(3):参照实施例1的步骤(1)-(3);
[0144]
(4)4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯的制备:
[0145]
取1,3-二甲基-1h-吡唑-4-甲酸(0.39g,2.81mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.90g,3.51mmol)与三乙胺(0.28g,2.81mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰(1.00g,3.51mmol)与三乙胺(0.28g,2.81mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯0.37g,收率64.76%。
[0146]
实施例14:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯(即化合物z
14
)的制备方法,包括以下步骤:
[0147]
步骤(1)-(3):参照实施例8的步骤(1)-(3);
[0148]
(4)4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯的制备:
[0149]
取1-甲基-3-三氟甲基吡唑-4-甲酸(0.26g,1.35mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.43g,1.69mmol)与三乙胺(0.14g,1.35mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-硝基-5-氯苯甲酰(0.50g,1.69mmol)与三乙胺(0.14g,1.35mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯0.18g,收率56.41%。
[0150]
实施例15:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯,包括以下步骤:
[0151]
步骤(1)-(3):参照实施例2的步骤(1)-(3);
[0152]
(4)4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯的制备:
[0153]
取环丙基异噁唑甲酸(0.37g,2.43mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.78g,3.04mmol)与三乙胺(0.25g,2.43mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氯-4-甲砜基苯甲酰(0.50g,1.52mmol)与三乙胺(0.25g,2.43mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯0.17g,收率60.24%。
[0154]
实施例16:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯(即化合物z
16
)的制备方法,包括以下步骤:
[0155]
步骤(1)-(3):参照实施例10的步骤(1)-(3);
[0156]
(4)4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯的制备:
[0157]
取1-甲基-3-三氟甲基吡唑-4-甲酸(0.29g,1.49mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.48g,1.86mmol)与三乙胺(0.15g,1.49mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氟-4-氯苯甲酰(0.50g,1.86mmol)与三乙胺(0.15g,1.49mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯0.11g,收率33.22%。
[0158]
实施例17:4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯(即化合物z
17
)的制备方法,包括以下步骤:
[0159]
步骤(1)-(3):参照实施例8的步骤(1)-(3);
[0160]
(4)4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯的制备:
[0161]
取1,3-二甲基吡唑-4-甲酸(0.23g,1.62mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.52g,2.03mmol)与三乙胺(0.16g,1.62mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-硝基-5-氯苯甲酰(0.60g,2.03mmol)与三乙胺(0.16g,1.62mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-硝基-5-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯0.24g,收率70.77%。
[0162]
实施例18:4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯(即化合物z
18
)的制备方法,包括以下步骤:
[0163]
步骤(1)-(3):参照实施例10的步骤(1)-(3);
[0164]
(4)4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯的制备:
[0165]
取环丙基异噁唑甲酸(0.64g,4.17mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(1.33g,5.21mmol)与三乙胺(0.42g,4.17mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氟-4-氯苯甲酰(0.70g,2.61mmol)与三乙胺(0.42g,4.17mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-氟-4-氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-5-环丙基异噁唑-4-羧酸酯0.19g,收率45.15%。
[0166]
实施例19:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯(即化合物z
19
)的制备方法,包括以下步骤:
[0167]
步骤(1)-(3):参照实施例1的步骤(1)-(3);
[0168]
(4)4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯的制备:
[0169]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰(0.50g,1.75mmol)与碳酸钾(0.36g,2.63mmol)在乙腈溶液(10ml)中混匀,在室温下加入4-硝基苯磺酰氯(0.78g,3.51mmol)与碘化钾(0.03g,0.18mmol),将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-4-硝基苯磺酸酯0.44g,收率53.35%。
[0170]
实施例20:4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯(即化合物z
20
)的制备方法,包括以下步骤:
[0171]
步骤(1)-(3):参照实施例2的步骤(1)-(3);
[0172]
(4)4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯的制备:
[0173]
取1,3-二甲基吡唑-4-甲酸(0.27g,1.90mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.39g,1.52mmol)与三乙胺(0.19g,1.90mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氯-4-甲砜基苯甲酰(0.50g,1.52mmol)与三乙胺(0.19g,1.90mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1,3-二甲基-1h-吡唑-4-羧酸酯0.29g,收率83.88%。
[0174]
实施例21:1-((4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯(即化合物z
21
)的制备方法,包括以下步骤:
[0175]
步骤(1)-(3):参照实施例2的步骤(1)-(3);
[0176]
(4)1-((4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯的制备:
[0177]
取1,3-二甲基-5-羟基-1h-吡唑-4-基-2-氯-4-甲砜基苯甲酰(0.50g,1.52mmol)与碳酸钾(0.32g,2.28mmol)在乙腈溶液(10ml)中混匀,在室温下加入乙基异丙基碳酰氯(0.46g,3.04mmol)与碘化钾(0.03g,0.15mmol),将反应体系升温至85℃进行回流反应,反应完全后,体系经减压浓缩、萃取及柱层析纯化后得到1-((4-(2-氯-4-甲砜基苯甲酰基)-1,3-二甲基-1h-吡唑-5-基)羟基)乙基异丙基碳酸酯0.41g,收率60.60%。
[0178]
实施例22:4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯(即化合物z
22
)的制备方法,包括以下步骤:
[0179]
步骤(1)-(3):参照实施例1的步骤(1)-(3);
[0180]
(4)4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯的制备:
[0181]
取1-甲基-3-三氟甲基吡唑-4-甲酸(0.44g,2.24mmol)与二氯甲烷(10ml)混匀,在室温下加入cmpi(0.72g,2.81mmol)与三乙胺(0.23g,2.24mmol),搅拌一段时间后再加入1,3-二甲基-5-羟基-1h-吡唑-4-基-2,4-二氯苯甲酰(0.80g,2.81mmol)与三乙胺(0.23g,2.24mmol),反应体系在室温条件下进行反应,反应完全后,体系经饱和碳酸氢钠溶液洗涤、干燥、减压浓缩,重结晶及柱层析纯化后得到4-(2,4-二氯苯甲酰基)-1,3-二甲基-1h-吡唑-5-基-1-甲基-3-三氟甲基-1h-吡唑-4-羧酸酯0.29g,收率56.02%。
[0182]
以上实施例制得的目标化合物的结构式及分子式如表1所示,其理化性质及谱图信息如表2所示;
[0183]
表1实施例1-22制得的目标化合物的分子式及结构式
[0184]
[0185]
[0186]
[0187][0188]
表2实施例1-22制得的目标化合物的理化性质和波谱数据
[0189]
[0190]
[0191]
[0192][0193]
目标化合物抑制靶标酶hppd的活性测试:
[0194]
本试验采用拟南芥对羟基苯丙酮酸双加氧酶(hppd)酶联免疫分析试剂盒进行测定化合物对hppd的体外抑制作用。苯唑草酮和硝磺草酮作为阳性对照。所有化合物用dmso溶解,实验操作严格按照试剂盒说明书进行,分别设空白孔(空白对照孔不加样品及酶标试剂,其余各步操作相同)、标准孔、待测样品孔。在酶标包被板上标准品准确加样50μl,待测样品孔中先加样品稀释液40μl,然后再加待测样品10μl(样品最终稀释度为5倍)。将样品加于酶标板孔底部,尽量不触及孔壁,轻轻晃动混匀。用封板膜封板后置37℃温育30分钟。小心揭掉封板膜,弃去液体,甩干,每孔加满洗涤液,静置30秒后弃去,如此重复5次,拍干后在每个孔中加入酶标试剂50μl,空白孔除外。重复温育和洗涤后每孔先加入显色剂a 50μl,在加入显色剂b 50μl,轻轻震荡混匀,37℃避光显色10分钟。而后加终止液50μl终止反应。以空白孔调零,在450nm波长下用酶标仪依次测量各孔的吸光度(od值),并根据以标准物质浓度为横坐标、od值为纵坐标绘制的标准曲线计算出半数抑制浓度(ic
50
)。
[0195]
目标化合物苗前活性测试:
[0196]
本试验采用培养皿法,以多种杂草作为待测对象,将杂草种子在培养箱条件下催芽至露白,而后在培养皿(12孔板)内铺2张滤纸,每皿(每孔)内摆放大小一致的露白后种子4-6粒,将化合物用dmf溶解后配制成100μg/ml浓度的药剂,后往孔中添加药剂,每处理重复三次,处理完毕后将培养皿置于人工气候培养箱,在25-30℃、光照5000lx、光照周期为昼:夜=16:8、相对湿度70%-80%的条件下培养5天,测量长势基本一致的4粒种子的根长和茎长,分别计算各处理组对杂草种子的生长抑制率,用l表示马齿苋根长及茎长,公式如下:
[0197]
生长抑制率(%)=l
(空白组)
–
l
(处理组)
/l
(空白组)
×
100%
[0198]
目标化合物苗后活性测试:
[0199]
本试验采用喷雾法,以多种杂草作为待测对象,将杂草种子在培养箱条件下催芽至露白,所有露白的杂草种子被均匀地撒在8x 8cm装中有三分之二有机基质土的塑料花盆里,并放在温室中生长。当禾本科杂草和阔叶杂草皆生长到三叶期时,可用于测试。化合物用100μldmf溶解,用0.1%tween-80稀释至150或37.5g ai ha的剂量。苯唑草酮和硝磺草酮作为阳性对照和目标化合物对所有杂草进行药物喷洒处理。经处理后的杂草,在温室内放置15天后,用目测法对照ck组评价其除草活性。作物安全性测试亦使用该苗后测试方法。
[0200]
按上述方法测定实施例1-22的目标化合物的除草活性及hppd酶抑制ic
50
值,结果见表3-7所示;
[0201]
表3实施例1-22目标化合物的hppd酶抑制活性
[0202][0203]
表4实施例1-22目标化合物在100μg/ml的浓度下的苗前除草活性
[0204]
[0205][0206]
表5实施例1-22目标化合物在150及37.5g ai ha的剂量下的苗后除草活性
[0207]
[0208][0209]
表6实施例5、15、20、21目标化合物在150g ai ha的剂量下的苗后除草活性
[0210][0211]
表7实施例5、15、20、21目标化合物在150g ai ha的剂量下的作物安全性
[0212][0213]
缩写:苯唑和硝磺分别代表苯唑草酮和硝磺草酮;cs、lp、po、cd、bn、vp、amt、sn、ce、tm、sj、tr、ma、lc、ei、sv分别代表小藜、黑麦草、马齿苋、狗牙根、油菜、婆婆纳、苋菜、龙葵、苦麦菜、蒲公英、鼠尾草、三叶草、繁缕、千金子、牛筋草、狗尾草
[0214]
由表3-7可以看出,化合物z6、z9、z
16
和z
21
对hppd的抑制活性最好,ic
50
值分别为0.08μm、0.05μm、0.08μm和0.11μm,优于苯唑草酮(1.33μm)和硝磺草酮(1.76μm)。苗前除草活性测试结果表明化合物z1、z9、z
10
、z
11
、z
18
和z
22
在100μg/ml的浓度下对苋菜根茎的抑制率均为100%,抑制效果均优于苯唑草酮(34.8%和40.2%)和硝磺草酮(14.4%和30.4%)。在100μg/ml的药物浓度下,化合物z
21
对稗草茎抑制率为44.3%,对其根抑制率为69.6%,分别优于苯唑草酮(16.0%和53.0%)和硝磺草酮(12.8%和41.7%)。苗后除草活性测试结果表明大多数吡唑化合物在剂量为150g ai ha时,对苘麻具有良好的除草活性,抑制率均在80%以上。化合物z5、z
15
、z
20
和z
21
对稗草和马唐具有优秀的除草活性,在150g ai ha时抑制率达到100%,且杂草完全被漂白,其生长受到严重抑制。同时,化合物z5、z
15
、z
20
和z
21
在37.5g ai ha的剂量下对稗草的抑制率为100%,与苯唑草酮(100%)一致,但优于硝磺草酮(80%)。选择具有较好苗后除草活性的化合物z5、z
15
、z
20
和z
21
进一步研究发现这些化合物具有和苯唑草酮相近的广谱性,特别是化合物z
21
对油菜、马齿苋、千金子及牛筋草显示出优秀的除草活性。同时,这些化合物对玉米、大豆、棉花、小麦和水稻等作物显现出优于苯唑草酮的安全性。
[0215]
综上所述,该系列吡唑衍生物具有优异的除草及酶抑制活性,其中,化合物z
21
不仅具有良好的hppd抑制作用,ic
50
值为0.11μm,而且具有优异的除草活性和作物安全性。因此,该系列化合物可作为新型的hppd抑制剂、苗前和苗后除草剂,被进一步开发从而能应用于更多的农田。
[0216]
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,任何未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1