一种铜绿假单胞菌O10血清型O-抗原三糖及其合成方法
一种铜绿假单胞菌o10血清型o-抗原三糖及其合成方法
技术领域
1.本发明属于化学合成领域,具体涉及一种铜绿假单胞菌o10血清型o-抗原三糖及其合成方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.铜绿假单胞菌(pseudomonas aeruginosa)是一种革兰氏阴性菌,广泛存在于自然界中,是临床最常见的条件性致病菌之一,常引起呼吸道、尿路、伤口和烧伤等多种感染(fems microbiol.rev.2021,45,1-20.)。呼吸道感染是铜绿假单胞菌引起的常见健康问题,据报道,2021年在我国医院获得性肺炎病原谱中,铜绿假单胞菌约占16%-22%,位列第二(chin.j.tubere.respir.dis.2022,45,739-752.)。机械通气患者、免疫功能低下患者、恶性肿瘤或hiv感染患者是该菌的易感人群(am.j.resp.crit.care.2005,171,1209-1214.)。铜绿假单胞菌具有严重的抗生素耐药性(j.infect.dis.2008,197,1079-1081.),根据《柳叶刀》公布的数据,2019年可归因于铜绿假单胞菌抗生素耐药性并与之相关的全球死亡病例超过25万例(lancet.2022,399,629-655.)。国内细菌耐药监测网的数据显示,2021年所有临床分离菌株中,铜绿假单胞菌的占比约为8%,居于第四位。此外,该菌对常用抗菌药物的耐药率也在不断攀升,对替卡西林钠克拉维酸的耐药率已达到30%(chinet)。面对铜绿假单胞菌耐药率的不断上升趋势,开发一种安全有效的疫苗以防治铜绿假单胞菌感染是更加切实可行的策略。
4.脂多糖(lps)是铜绿假单胞菌重要的毒力因子之一,其结构中的o-抗原寡糖由一些在人体中不常见的稀有单糖构成(surf.interfaces.2021,26,101415-101425.)。o-抗原具有一定的免疫原性,是研发相关疫苗的理想靶标抗原。根据o-抗原的结构及生物学特异性,铜绿假单胞菌可分为20种血清型,其中一个重要血清型o10的o-抗原寡糖由[
→
4)-α-l-乙酰氨基半乳糖醛酸-(1
→
3)-α-d-乙酰氨基喹诺糖-(1
→
3)-α-l-鼠李糖-(1
→
]n三糖重复片段组成。该o10血清型o-抗原寡糖包含两个亚型o10
a,b
和o10
a,c
,两者区别在于l-鼠李糖砌块c2-oh有无乙酰基(eur.j.biochem.1982,125,221-227.)。
[0005]
从天然菌体中提取o-抗原寡糖的纯化步骤较为繁琐复杂,产品质量难以控制,并可能会掺杂其他细菌毒力因子,进而导致后续制备的糖疫苗可能发生严重的副作用(chem.biol.2014,21,38-50.)。利用化学合成的方法来制备o-抗原寡糖,可避免生物提取方法存在的抗原结构不均一以及掺杂其他未知毒力因子的隐患。此外,结构均一的合成寡糖抗原便于后续抗原结构的优化和构效关系研究,有利于疗效更佳、质量更加可控、副作用更少的新型半合成或全合成糖缀合物疫苗研发。
[0006]
糖类化合物具有结构复杂性和多样性,其立体化学合成难度较高。在铜绿假单胞菌o10血清型o-抗原三糖的合成中,稀有寡糖模块的高效制备、1,2-顺式和1,2-反式糖苷键
的立体选择性构建、特定位置官能团的引入与修饰是其化学合成的关键。目前还没有铜绿假单胞菌o10血清型o-抗原三糖的合成报导。
技术实现要素:
[0007]
为了解决现有技术的不足,本发明的目的是提供一种铜绿假单胞菌o10血清型o-抗原三糖及其合成方法。本发明通过三个单糖砌块化学合成铜绿假单胞菌o10血清型的o-抗原三糖,合成砌块包括利用l-半乳糖胺、d-喹诺糖胺和l-鼠李糖砌块,单糖砌块之间通过1,2-顺式或1,2-反式糖苷键连接。首先,单糖砌块之间通过立体选择性糖苷化偶联组装三糖骨架,随后通过官能团修饰和保护基脱除等步骤得到目标三糖。同时,三糖的还原端连接臂末端组装有自由氨基,为下一步的缀合免疫原性载体做准备。
[0008]
为了实现上述目的,本发明是通过如下的技术方案来实现:
[0009]
第一方面,本发明提供了一种铜绿假单胞菌的o10血清型o-抗原三糖的合成方法,包括:利用l-半乳糖胺砌块、d-喹诺糖胺砌块和l-鼠李糖砌块合成铜绿假单胞菌o10血清型o-抗原三糖,单糖砌块之间通过α-1,2-顺式或1,2-反式糖苷键连接。
[0010]
第二方面,本发明提供了一种铜绿假单胞菌的o10血清型o-抗原三糖,由上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到。
[0011]
第三方面,本发明提供了一种糖缀合物,利用上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到的产品和/或上述铜绿假单胞菌的o10血清型o-抗原三糖作为原料制备得到。
[0012]
第四方面,本发明提供了上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到的产品和/或上述o10血清型o-抗原三糖和/或上述糖缀合物在制备抗铜绿假单胞菌疫苗以及在治疗铜绿假单胞菌感染的药物中的应用。
[0013]
上述本发明的一种或多种技术方案取得的有益效果如下:
[0014]
1.本发明提出了一种铜绿假单胞菌o10血清型o-抗原三糖的化学合成方法,通过对稀有寡糖模块的合成、糖苷键的立体选择性构建、特定官能团的位置选择性修饰以及脱除保护基等步骤成功完成了铜绿假单胞菌o10血清型两个o-抗原三糖的化学合成。
[0015]
2.本发明合成的铜绿假单胞菌o10血清型o-抗原三糖的还原端位置组装有氨基连接臂,可以和免疫原性载体(如蛋白质)缀合以制备糖缀合物疫苗,对预防和治疗铜绿假单胞菌感染具有重要意义。
附图说明
[0016]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0017]
图1为l-半乳糖胺砌块3的合成路线图;
[0018]
图2为d-喹诺糖胺砌块6的合成路线图;
[0019]
图3为l-鼠李糖砌块9的合成路线图;
[0020]
图4为二糖化合物11的合成路线图;
[0021]
图5为三糖16和17的合成路线图。
具体实施方式
[0022]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0023]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0024]
本发明的一种典型具体实施方式中,提供了一种铜绿假单胞菌的o10血清型o-抗原三糖的合成方法,包括:利用l-半乳糖胺砌块、d-喹诺糖胺砌块和l-鼠李糖砌块合成铜绿假单胞菌o10血清型o-抗原三糖,单糖砌块之间通过α-1,2-顺式或1,2-反式糖苷键连接。
[0025]
本发明的又一种典型具体实施方式中,所述l-半乳糖胺砌块的化学结构如式i所示,d-喹诺糖胺砌块的化学结构式如式ii所示,l-鼠李糖砌块的化学结构式如式iii所示;
[0026][0027]
其中,linker为-(ch2)
n-n-y1y2、-(ch2)
n-s-y1y2和-(ch2)
n-n3中的一种,n=2-40,y1为氢或者苄基,y2为氢或者苄氧羰基;
[0028]
r1,r2,r3,r4包括氢、氮或者乙酰基;
[0029]
r5,r6,r8,r
10
为氢、酯基或者醚基,包括但不限于氢、乙酰基、新戊酰基、苯甲酰基、氯乙酰基、乙酰丙酰基、烯丙氧羰酰基、苄基、对甲氧基苄基、2-萘甲基、烯丙基、三苯甲基;
[0030]
r7为氢、酯基或者醚基包括但不限于氢、乙酰基、苯甲酰基、新戊酰基、氯乙酰基、乙酰丙酰基、烯丙氧羰酰基、苄基、2-萘甲基、对甲氧基苄基、烯丙基、三苯甲基、叔丁基二甲基硅基、叔丁基二苯基硅烷基、三乙基硅烷基;
[0031]
r9为氢或者酯基包括但不限于氢、乙酰基、新戊酰基、苯甲酰基、氯乙酰基、乙酰丙酰基、烯丙氧羰酰基;
[0032]
lg包括硫基、酯基、硒苯基、卤素、亚胺酯基、膦酸酯基、醚基但不限于乙硫基、对甲苯硫基、苯硫基、乙酰基、氟、氯、溴、三氯乙酰亚氨酯、n-苯基三氟乙酰亚胺酯、二丁基磷酸酯、4-戊烯基氧基。
[0033]
本发明的又一种典型具体实施方式中,所述合成方法包括利用d-喹诺糖胺砌块和l-鼠李糖砌块合成二糖片段,所述二糖片段的化学结构如式iv所示,
[0034][0035]
本发明的又一种典型具体实施方式中,所属合成方法包括利用二糖片段与l-半乳
糖胺砌块合成三糖片段,所述三糖片段化学结构式如式v所示,
[0036][0037]
本发明的又一种典型具体实施方式中,所述合成方法包括将合成的三糖片段中l-半乳糖胺的c6-oh氧化成羧酸,进而得到的化合物化学结构式如式vi所示,
[0038][0039]r11
为氢、醚基包括但不限于甲基、苄基、对甲氧基苄基、2-萘甲基、三苯甲基等。
[0040]
本发明的又一种典型具体实施方式中,所述l-半乳糖胺砌块的合成方法包括:以硒苯基2-叠氮-2-去氧-α-l-半乳糖苷为起始原料,c6-oh选择性用叔丁基二甲基硅基保护,c3-oh和c4-oh采用苄基保护。
[0041]
本发明的又一种典型具体实施方式中,所述d-喹诺糖胺砌块的合成方法包括:以对甲苯硫基4-苄基-β-d-鼠李糖苷为起始原料,对c3-oh进行位置选择性保护,剩余c2-oh进行叠氮基转位得d-喹诺糖胺砌块;
[0042]
本发明的又一种典型具体实施方式中,所述l-鼠李糖砌块的合成方法包括:在l-鼠李糖还原端组装氨基连接臂,最后脱除对甲氧基苄基保护基得l-鼠李糖砌块。
[0043]
本发明的又一种典型具体实施方式中,提供了一种铜绿假单胞菌的o10血清型o-抗原三糖,利用上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到。
[0044]
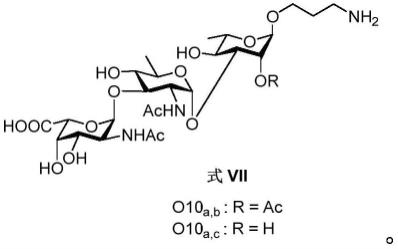
本发明的又一种典型具体实施方式中,所述铜绿假单胞菌的o10血清型o-抗原三糖为o10
a,b
或o10
a,c
,化学式结构如式vii所示,
[0045][0046]
本发明的又一种典型具体实施方式中,提供了一种糖缀合物,利用上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到的产品和/或上述铜绿假单胞菌的o10血清型o-抗原三糖作为原料制备得到。
[0047]
本发明的又一种典型具体实施方式中,提供了上述铜绿假单胞菌的o10血清型o-抗原三糖的合成方法合成得到的产品和/或上述铜绿假单胞菌的o10血清型o-抗原三糖和/
或上述糖缀合物在制备抗铜绿假单胞菌疫苗以及在治疗铜绿假单胞菌感染的药物中的应用。
[0048]
为了充分说明本发明的制备思路,下面结合具体实施例对本方案的过程进行描述,实施例仅供举例说明,不应被解释或理解为对本发明保护限制,此外,本发明所有试验材料如无特殊说明均为市售购买产品。本发明的产率计算方法为“产物(mol)/反应底物(mol)*100%”。本发明中化合物结构鉴定的方法为核磁共振图谱的测定(安捷伦,600mhz),高分辨质谱测定结果在每个化合物的具体合成中列出。
[0049]
实施例1
[0050]
l-半乳糖胺砌块3的合成:
[0051]
如图1所示,以硒苯基2-叠氮-2-去氧-α-l-半乳糖苷化合物1(chem.eur.j.2005,11,1010-1016.)为起始原料,吡啶为溶剂,c6-oh选择性用叔丁基二甲基硅基保护得化合物2。随后,以无水n,n-二甲基甲酰胺(dmf)为溶剂,冰浴条件下,2与氢化钠和溴化苄反应,得化合物3。
[0052]
具体实验操作和步骤:
[0053]
化合物2:在氮气保护条件下,将硒苯基2-叠氮-2-去氧-α-l-半乳糖苷化合物1(500.0mg,1.50mmol)溶解于吡啶(5.0ml)中,冰浴条件下加入叔丁基二甲基氯硅烷(350.0mg,2.40mmol)。反应体系逐渐升至室温,并搅拌10小时。tlc检测原料点反应完全。向体系加入二氯甲烷(30.0ml)稀释,用饱和食盐水洗涤,干燥有机相,减压蒸干。将粗产物用硅胶柱分离纯化(二氯甲烷:乙酸乙酯=24:1)得到白色浆状化合物2(642.6mg,93%)。
[0054]
化合物3:在氮气保护条件下,用无水dmf(5.0ml)溶解化合物2(600.0mg,1.30mmol),冰浴条件下依次加入60%的氢化钠(157.0mg,3.90mmol)和溴化苄(0.47ml,3.90mmol)。反应0.5小时后,tlc检测到原料反应完全。冰浴条件下,向体系中滴加适量冰水淬灭反应,用乙酸乙酯(30.0ml)稀释,随后用饱和食盐水洗涤。有机相用无水硫酸钠干燥后减压旋干,所得粗产物经硅胶柱分离纯化(石油醚:乙酸乙酯=50:1),得白色浆状化合物3(756.0mg,91%)。1hnmr(600mhz,cdcl3)δ:7.50-7.75(m,4h),7.35-7.45(m,8h),7.24-7.22(m,1h),5.92(d,j=5.4hz,1h),4.78(d,j=11.4hz,1h),4.76(d,j=11.4hz,1h),4.74(d,j=11.4hz,1h),4.58(d,j=11.4hz,1h),4.34(dd,j=5.4,10.2hz,1h),4.15-4.39(m,1h),4.04-3.99(m,1h),3.73(dd,j=3.0,10.2hz,1h),3.69(dd,j=7.8,9.6hz,1h),3.48(dd,j=6.0,10.2hz,1h),0.88(s,9h),0.02(s,3h),0.01(s,3h).
13
c nmr(150mhz,cdcl3)δ:138.4,137.5,134.6,129.0,128.6,128.5,128.0,127.9,127.7,127.6,85.5,80.3,75.0,73.5,72.9,72.5,61.2,60.9,25.9,18.2,-5.4,-5.5.hr esi-tof ms(m/z):calcd for c
32h41
n3o4sesina[m+na]
+
,662.1924;found,662.1948。
[0055]
实施例2
[0056]
d-喹诺糖胺砌块6的合成如图2:
[0057]
如图2所示,以化合物4为起始原料,在二丁基氧化锡和4-甲氧基苄基氯的作用下,对其c3-oh进行位置选择性保护,得到化合物5,剩余c2-oh进行叠氮转位得d-喹诺糖胺砌块6。
[0058]
具体实验操作和步骤:
[0059]
化合物5:将化合物4(600.0mg,1.67mmol)和二丁基氧化锡(497.8mg,2.00mmol)溶
解于甲苯(30.0ml)中,安装分水器和冷凝管,加热回流8小时。随后冷却至室温,减压旋蒸除去甲苯,真空干燥1小时。在氮气保护下,加入无水dmf(8.0ml)和氟化铯(381.3mg,2.51mmol),随后在冰浴条件下加入四丁基溴化铵(809.1mg,2.51mmol)和4-甲氧基苄氯(0.50ml,3.34mmol)。反应体系自然升至室温,搅拌过夜。tlc检测直至原料反应完全。向反应体系中加入乙酸乙酯(30.0ml)稀释,然后用饱和食盐水洗涤,用无水硫酸钠干燥有机相,减压旋干得到粗产物。粗品经硅胶柱分离纯化(石油醚:乙酸乙酯=6:1)得到白色浆状化合物5(690.0mg,87%)。1hnmr(600mhz,cdcl3)δ:7.43-7.39(m,2h),7.36-7.27(m,6h),7.26-7.25(m,1h),7.12-7.09(m,2h),6.89-6.84(m,2h),4.88(d,j=11.4hz,1h),4.68(t,j=1.2hz,1h),4.66-4.62(m,2h),4.60(d,j=10.8hz,1h),4.24-4.22(m,1h),3.80(s,3h),3.54(dd,j=3.6,9.0hz,1h),3.49(t,j=9.0hz,1h),3.35-3.28(m,1h),2.64(dd,j=1.2,2.4hz,1h),2.33(s,3h),1.35(d,j=6.0hz,3h).
13
c nmr(150mhz,cdcl3)δ:159.5,138.2,137.5,131.1,129.7,129.7,129.62,129.59,128.4,128.1,127.8,114.0,86.82,82.13,79.4,75.9,75.5,71.5,70.1,55.3,21.1,18.2.hr esi-tof ms(m/z):calcd for c
28h32
o5sna[m+na]
+
,503.1863;found,503.1876。
[0060]
化合物6:在氮气保护条件下,将化合物5(650.0mg,1.35mmol)溶解于无水二氯甲烷(4.0ml),加入吡啶(0.87ml,10.80mmol)。在冰盐浴中,缓慢滴加三氟甲磺酸酐(0.77ml,5.40mmol),搅拌0.5小时后,tlc检测原料反应完全,加入饱和碳酸氢钠溶液,直至无气泡产生。加入二氯甲烷稀释,用饱和食盐水洗涤,干燥有机相,减压蒸馏得到粗品。粗品经硅胶柱分离纯化(石油醚:乙酸乙酯=10:1)得到三氟甲磺酸酯中间体。在氮气保护条件下,将生成的中间体溶解在无水dmf(4.0ml)中,冰浴条件下加入叠氮化钠(702.0mg,10.80mmol),室温下搅拌2小时后,tlc检测到原料反应完全。向体系中加入乙酸乙酯(40.0ml)稀释,用饱和食盐水洗涤,有机相干燥后减压旋干,所得粗产物用硅胶柱分离纯化(石油醚:二氯甲烷=1:1.5),得到白色浆状化合物6(572.7mg,两步反应收率84%)。1h nmr(600mhz,cdcl3)δ:7.49-7.44(m,2h),7.37-7.28(m,5h),7.26-7.24(m,2h),7.16-7.10(m,2h),6.90-6.79(m,2h),4.84(d,j=10.8hz,1h),4.78(d,j=9.6hz,1h),4.75(d,j=10.2hz,1h),4.63(d,j=10.8hz,1h),4.32(d,j=10.2hz,1h),3.78(s,3h),3.45(t,j=9.6hz,1h),3.41-3.35(m,1h),3.26(dd,j=9.0,9.6hz,1h),3.09(t,j=9.6hz,1h),2.34(s,3h),1.32(d,j=6.0hz,3h).
13
c nmr(150mhz,cdcl3)δ:159.5,138.7,137.9,134.2,130.0,129.77,129.75,128.5,127.9,127.8,127.2,113.9,85.9,84.6,83.2,75.9,75.5,75.3,65.3,55.3,21.2,18.1.hr esi-tof ms(m/z):calcd for c
28h31
n3o4sna[m+na]
+
,528.1927;found,528.1919。
[0061]
实施例3
[0062]
l-鼠李糖砌块9的合成如图3:
[0063]
如图3所示,以二氯甲烷为溶剂,nis(碘代丁二酰亚胺)和tfoh(三氟甲磺酸)为活化体系,冰浴条件下,化合物7和n-苄基-n-甲酸苄酯-3-氨基丙醇进行糖苷化反应得化合物8。将化合物8溶于甲苯中,冰浴条件下利用10% tfa-甲苯溶液脱除3-o-位的对甲氧基苄基得2-o-位乙酰基保护的l-鼠李糖砌块9。
[0064]
具体实验操作和步骤:
[0065]
化合物8:在氮气保护条件下,将化合物7(500.0mg,0.96mmol)、n-苄基-n-甲酸苄酯-3-氨基丙醇(257.0mg,0.86mmol)和分子筛溶于无水二氯甲烷(4.0ml)中,室温下干
燥1小时后,冰浴条件下,依次加入nis(260.0mg,1.15mmol)和tfoh(8.4μl,0.01mmol),继续搅拌15分钟后,tlc检测到反应完全。向反应体系内滴加适量的三乙胺淬灭,抽滤除去分子筛,加入二氯甲烷(30.0ml)稀释,依次用饱和硫代硫酸钠溶液、饱和碳酸氢钠溶液和饱和食盐水洗涤。有机相用无水硫酸钠干燥后减压旋干,所得的粗产物用硅胶柱分离纯化(石油醚:乙酸乙酯=4:1)得到白色泡沫状化合物8(533.5mg,89%)。1h nmr(600mhz,cdcl3)δ:7.40-7.26(m,13h),7.25-7.15(m,4h),6.84-6.78(m,2h),5.30(s,1h),5.18(d,j=19.8hz,2h),4.90(d,j=10.8hz,1h),4.65-4.47(m,5h),4.44-4.34(m,1h),3.88-3.81(m,1h),3.76(s,3h),3.70-3.57(m,2h),3.41-3.27(m,4h),3.14(s,3h),1.85-1.71(m,2h),1.29(d,j=6.0hz,3h).
13
c nmr(150mhz,cdcl3)δ:170.4,159.2,156.7,156.0,138.5,137.8,136.8,136.7,130.1,129.7,128.6,128.51,128.47,128.4,128.3,127.9,127.8,127.73,127.66,127.4,127.3,113.8,97.7,77.7,76.8,75.4,71.4,69.0,67.7,67.2,65.3,55.2,50.7,44.1,28.0,21.14,17.98.hr esi-tof ms(m/z):calcd for c
41h47
no9na[m+na]
+
,720.3143;found,720.3132。
[0066]
化合物9:将化合物8(560.0mg,0.80mmol)溶于甲苯(3.0ml)中,冰浴条件下缓慢滴加90% tfa水溶液(0.4ml),搅拌2小时后,tlc检测到原料反应完全。加入饱和碳酸氢钠溶液,直至无气泡产生。向体系中加入乙酸乙酯(30.0ml)稀释,用饱和食盐水(10.0ml
×
3)洗涤,干燥有机相,减压蒸馏得到粗品。粗品经硅胶柱分离纯化(石油醚:乙酸乙酯=2:1)得到白色泡沫化合物9(400.0mg,88%)。
[0067]
实施例4
[0068]
二糖化合物11的合成如图4:
[0069]
如图4所示,二糖10由通过图2路线合成的d-喹诺糖胺砌块6和由图3路线合成的l-鼠李糖衍生物9经糖苷化偶联制得。在二氯甲烷和乙醚混合溶剂中,nis和tfoh为活化剂,冰浴条件下,反应生成α和β构型混合的二糖10。随后,以甲苯为溶剂,利用10% tfa-甲苯溶液脱除3-o-对甲氧基苄基,分离纯化得到α构型的二糖11。
[0070]
具体实验操作和步骤:
[0071]
化合物11:在氮气保护条件下,将化合物6(420.0mg,0.83mmol)、化合物9(400.0mg,0.70mmol)和分子筛溶解于无水二氯甲烷和乙醚混合溶剂(5.0ml,v:v=1:1)中,室温下干燥1小时后,冰浴条件下,依次加入nis(224.0mg,1.00mmol)和tfoh(7.3μl,0.08mmol),继续搅拌15分钟后,tlc检测到反应完全。向反应体系内滴加适量的三乙胺淬灭,抽滤除去分子筛,加入二氯甲烷(40.0ml)稀释,依次用饱和硫代硫酸钠溶液、饱和碳酸氢钠溶液和饱和食盐水洗涤。有机相用无水硫酸钠干燥后减压旋干,所得粗产物用硅胶柱分离纯化(石油醚:乙酸乙酯=6:1)得到白色泡沫状化合物10(676.0mg,85%)。将化合物10(670.0mg,0.70mmol)溶于甲苯(3.0ml)中,冰浴条件下,缓慢滴加90% tfa的水溶液(0.4ml),2小时后,tlc检测到原料反应完全,加入饱和碳酸氢钠溶液,直至无气泡产生。向体系中加入乙酸乙酯(40.0ml)稀释,用饱和食盐水洗涤,干燥有机相,减压蒸馏得到粗品,然后用硅胶柱分离纯化(甲苯:乙酸乙酯=10:1)得到白色泡沫状的单一α-构型化合物11(258.0mg,44%)。1h nmr(600mhz,cdcl3)δ:7.38-7.26(m,14h),7.24-7.14(m,6h),5.29(s,1h),5.17(d,j=21.0hz,2h),5.02(d,j=3.6hz,1h),4.81(d,j=10.2hz,1h),4.75(d,j=11.4hz,1h),4.72(d,j=11.4hz,1h),4.64(s,1h),4.57(d,j=10.2hz,1h),4.51-4.45(m,
2h),4.16-4.09(m,2h),4.04-3.96(m,1h),3.76-3.55(m,2h),3.48(t,j=9.6hz,1h),3.41-3.23(m,4h),3.18-3.11(m,1h),3.09(t,j=9.6hz,1h),2.41(d,j=3.0hz,1h),2.20(s,3h),1.86-1.69(m,2h),1.31(d,j=6.0hz,3h),1.18(d,j=5.4hz,3h).
13
c nmr(150mhz,cdcl3)δ:170.5,138.2,137.7,137.5,136.7,128.60,128.57,128.5,128.4,128.0,127.8,127.7,127.4,127.3,97.42,92.97,84.7,79.5,76.1,75.2,72.4,67.8,67.1,66.9,65.6,65.4,62.8,50.8,50.6,44.5,43.6,28.2,27.7,20.8,18.0,17.8.hr esi-tof ms(m/z):calcd for c
46h54
n4o
11
k[m+k]
+
,877.3421;found,877.3429.
[0072]
实施例5
[0073]
三糖16和17的合成如图5:
[0074]
如图5所示,在二氯甲烷和乙醚混合溶剂中,以nis和tfoh为活化剂,在-10℃条件下,l-半乳糖胺砌块3与受体11进行糖苷化偶联,得到单一α-构型连接的三糖12。以氢氟酸吡啶络合物将三糖12上的叔丁基二甲基硅基脱除,得c6
″‑
oh化合物13。采用本课题组优化的2,2,6,6-四甲基哌啶氧化物(tempo)/碘苯二乙酸(baib)的氧化方法,完成糖醛酸的制备,随后将糖醛酸进行苄基化得化合物14。在1,3-丙二硫醇和三乙胺的作用下,将三糖14中的两个叠氮基还原成氨基,随后利用吡啶和醋酸酐将生成的氨基进行乙酰化得化合物15。最后,经氢氧化钯催化的氢解作用脱除15中所有苄基和苄氧羰基得到组装有氨基连接臂的铜绿假单胞菌o10a,b血清型o-抗原三糖16。随后利用甲醇钠溶液脱除16中的c2-o-ac得o10a,c血清型o-抗原三糖17。
[0075]
具体实验操作和步骤:
[0076]
化合物12:在氮气保护条件下,将l-半乳糖胺供体3(223.7mg,0.35mmol)、二糖受体11(120.0mg,0.14mmol)和分子筛溶解于无水二氯甲烷和乙醚的混合溶液(6.0ml,v:v=1:1)中,搅拌干燥1小时。将体系降温至-10℃,依次加入nis(94.5mg,0.42mmol)和tfoh(3.5μl,0.04mmol),搅拌0.5小时,tlc检测反应完全。向反应体系内滴加适量的三乙胺淬灭,抽滤除去分子筛,加入二氯甲烷(30.0ml)稀释,依次用饱和硫代硫酸钠溶液、饱和碳酸氢钠溶液和饱和食盐水洗涤。有机相用无水硫酸钠干燥后减压旋干,所得的粗产物用硅胶柱分离纯化(甲苯:乙酸乙酯=16:1)得到白色泡沫状化合物12(152.2mg,81%)。1h nmr(600mhz,cdcl3)δ:7.38-7.26(m,23h),7.24-7.12(m,7h),5.62(d,j=3.6hz,1h),5.28(s,1h),5.16(d,j=19.8hz,2h),5.05(d,j=3.6hz,1h),4.88(d,j=11.4hz,1h),4.83(d,j=11.4hz,1h),4.79(d,j=11.4hz,1h),4.66-4.52(m,6h),4.49-4.46(m,2h),4.21(dd,j=9.0,9.6hz,1h),4.14-4.10(m,1h),4.07-4.00(m,2h),3.98-3.96(m,1h),3.93(dd,j=3.0,10.8hz,1h),3.89(dd,j=2.4,10.8hz,1h),3.71-3.47(m,5h),3.36-3.17(m,5h),2.19(s,3h),1.82-1.70(m,2h),1.29(d,j=6.0hz,3h),1.10(d,j=4.2hz,3h),0.77(s,9h),-0.14(s,3h),-0.25(s,3h).
13
c nmr(150mhz,cdcl3)δ:170.5,156.1,138.6,138.1,137.7,137.6,137.3,128.9,128.6,128.49,128.46,128.2,128.0,127.9,127.8,127.5,127.3,97.4,96.9,93.2,83.4,79.2,76.2,75.4,74.8,74.0,73.1,72.5,71.9,71.5,67.7,67.2,67.0,66.8,65.5,62.7,60.6,59.9,50.7,44.0,27.9,25.9,20.8,18.0,17.9,17.8,-5.77,-5.80.hr esi-tof ms(m/z):calcd for c
72h93
n8o
15
si[m+nh4]
+
,1337.6524;found,1337.6585。
[0077]
化合物14:将化合物12(120.0mg,0.09mmol)溶解于二氯甲烷和乙腈混合溶液
(2.0ml,v:v=3:1)中,冰浴条件下逐滴加入氢氟酸吡啶络合物(0.20ml),室温搅拌1.5小时后,tlc检测反应底物12消失,逐滴加入淬灭剂饱和碳酸氢钠溶液,直到无气泡产生。以二氯甲烷(30.0ml)对反应体系进行稀释,用饱和食盐水溶液对有机相进行洗涤,有机相用无水硫酸钠干燥后减压旋干,所得的粗产物经硅胶柱分离纯化(石油醚:乙酸乙酯=4:1)得到白色泡沫状化合物13(96.4mg,86%)。在氮气保护条件下,将化合物13(90.0mg,0.07mmol)溶解于无水二氯甲烷(1.40ml)中,在冰浴条件下依次加入tempo(2.2mg,0.014mmol)和baib(34.0mg,0.11mmol),反应自然升至室温,搅拌4小时后,tlc检测反应完全。随后将反应体系移入冰浴中,依次加入水(14.0μl)和baib(68.0mg,0.22mmol),室温下搅拌20小时后,tlc检测到醛中间体消失。向反应体系中滴加适量的硫代硫酸钠溶液淬灭反应,加入二氯甲烷(20.0ml)稀释,依次用饱和碳酸氢钠溶液和饱和食盐水洗涤。有机相干燥后减压浓缩,并于油泵减压干燥2小时得粗产物,不经分离纯化,直接用于下步反应。将粗产物溶于无水dmf(3.0ml)中,氮气保护条件下,依次加入碳酸钾(19.0mg,0.14mmol)和溴化苄(13.0μl,0.11mmol)。反应搅拌0.5小时后,tlc检测反应完全,滴加冰水淬灭反应。以乙酸乙酯(30.0ml)稀释反应液,用饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸干,经硅胶柱分离纯化(石油醚:乙酸乙酯=7:1)得到白色泡沫状化合物14(76.1mg,两步反应收率83%)。1h nmr(600mhz,cdcl3)δ:7.37-7.26(m,14h),7.25-7.12(m,17h),7.09-7.06(m,2h),7.02-6.98(m,2h),5.62(d,j=3.6hz,1h),5.28(s,1h),5.17(d,j=21.0hz,2h),5.07(d,j=3.0hz,1h),4.77(d,j=10.2hz,1h),4.71(d,j=11.4hz,1h),4.69-4.58(m,7h),4.51-4.43(m,4h),4.28-4.22(m,2h),4.12-4.07(m,1h),4.06-4.01(m,2h),3.95(dd,j=3.6,10.2hz,1h),3.87(dd,j=2.4,10.8hz,1h),3.74-3.55(m,2h),3.47(t,j=9.6hz,1h),3.39-3.24(m,4h),3.47(t,j=9.6hz,1h),2.21(s,3h),1.84-1.69(m,2h),1.76(d,j=6.6hz,3h),1.14(s,3h).
13
c nmr(150mhz,cdcl3)δ:170.5,167.7,156.5,138.0,137.7,137.5,137.3,136.8,136.7,134.9,128.59,128.58,128.50,128.47,128.44,128.4,128.3,128.09,128.05,127.9,127.8,127.4,127.1,97.5,97.0,92.9,83.2,79.5,76.3,75.2,75.1,74.7,73.3,72.3,72.2,70.9,67.8,67.2,67.15,65.4,64.2,59.2,50.7,44.1,27.9,20.9,18.0,17.8.hr esi-tof ms(m/z):calcd for c
73h83
n8o
16
[m+nh4]
+
,1327.5922;found,1327.5925。
[0078]
化合物16:将化合物14(60.0mg,0.05mmol)溶于吡啶和水的混合溶液(1.50ml,v:v=4:1)中,室温下依次加入三乙胺(0.21ml,1.50mmol)和1,3-丙二硫醇(0.10ml,1.00mmol),搅拌2小时,tlc检测原料反应完全。将反应体系减压蒸干,真空干燥,得到粗产物,不经分离纯化,直接用于下步反应。用吡啶(2.00ml)溶解粗产物,加入醋酸酐(0.50ml),室温下反应1.5小时,tlc检测反应完全。将反应体系减压蒸馏旋干,所得粗产物经硅胶柱分离纯化(二氯甲烷:甲醇=30:1)得到白色泡沫状化合物15(56.0mg,83%)。将化合物15(44.0mg,0.016mmol)溶解于甲醇和水的混合溶液(7.0ml,v:v=6:1)中,滴加5滴冰醋酸,加入氢氧化钯(30.0mg),反应体系在50psi的氢气条件下搅拌48小时,经质谱检测,反应完全。随后,过滤除去不溶物,减压蒸馏除去溶剂,将所得粗产物在g-10葡聚糖凝胶柱中纯化,以水为流动相,浓缩含有样品的洗脱剂,得到无色透明浆状物16(18.0mg,85%)。1h nmr(600mhz,d2o)δ:5.30(dd,j=2.4,3.0hz,1h),5.15(d,j=4.2hz,1h),4.89(d,j=4.2hz,1h),4.85(d,j=1.8hz,1h),4.74(d,j=1.2hz,1h),4.30(dd,j=1.8,3.6hz,1h),4.14(dd,
j=3.6,10.8hz,1h),4.11(dd,j=4.2,10.8hz,1h),4.09
–
4.04(m,1h),3.96(dd,j=3.0,10.8hz,1h),3.94-3.90(m,2h),3.89-3.85(m,1h),3.80-3.75(m,1h),3.63(t,j=9.6hz,1h),3.61-3.60(m,1h),3.26(dd,j=9.0,9.6hz,1h),3.16-3.06(m,2h),2.20(s,3h),2.03(s,3h),2.01
–
1.98(m,2h),1.96(s,3h),1.36(d,j=6.0hz,3h),1.27(d,j=6.0hz,3h).
13
c nmr(150mhz,d2o)δ:176.0,174.2,173.3,172.6,97.2,96.6,92.9,74.2,73.9,71.9,71.8,69.9,68.9,67.70,67.67,67.4,65.1,53.6,49.4,37.4,26.6,22.1,22.0,20.4,16.7,16.6.hr esi-tof ms(m/z):calcd for c
27h46
n3o
16
[m+h]
+
,668.2873;found,668.2858。
[0079]
化合物17:将化合物16(10.0mg,0.015mmol)溶解在甲醇溶液(1.00ml)中,室温条件下,逐滴加入甲醇钠(0.5m的甲醇溶液)溶液,调节反应液ph值至10,搅拌1小时后,tlc检测原料反应完全,加入酸性树脂进行中和,过滤并减压浓缩得粗产物,将所得粗产物在g-10葡聚糖凝胶柱中纯化,水为流动相,浓缩含有样品的洗脱剂,得到无色透明浆状物17(8.6mg,92%)。1h nmr(600mhz,d2o)δ:5.17(d,j=4.2hz,1h),4.85(d,j=3.6hz,1h),4.82(d,j=1.2hz,1h),4.75(d,j=1.2hz,1h),4.31-4.29(m,1h),4.16-4.09(m,3h),4.03(dd,j=2.4,3.0hz,1h),4.00-3.94(m,2h),3.87-3.81(m,1h),3.73-3.67(m,2h),3.61-3.57(m,1h),3.55(t,j=9.6hz,1h),3.27(t,j=9.6hz,1h),3.13-3.04(m,2h),2.03(s,3h),1.99(s,3h),1.98-1.94(m,2h),1.33(d,j=6.0hz,3h),1.27(d,j=6.0hz,3h).
13
c nmr(150mhz,d2o)δ:176.0,174.2,173.3,99.4,96.7,94.7,76.0,74.4,73.9,70.0,69.9,68.7,68.0,67.6,67.0,65.0,53.9,49.4,37.4,27.0,22.1,21.8,16.7,16.6.hr esi-tof ms(m/z):calcd for c
25h44
n3o
15
[m+h]
+
,626.2767;found,626.2767。
[0080]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1