一种具有光热双重响应性的手性过盈螺烯类分子马达及其制备方法
1.本发明涉及多响应功能分子及其制备技术领域,具体涉及一种具有光热双重响应性的手性过盈螺烯类分子马达及其制备方法。
背景技术:
2.手性过盈螺烯类分子马达由荷兰科学家b.l.feringa课题组提出,并成功合成出不同结构的衍生物,因此而获得2016年诺贝尔化学奖。由于此类分子不仅具有一般的光响应分子所具有的光响应顺反异构,使得具有分子结构变化的功能,而且还具有绝大多数光响应分子所不具备的手性翻转的功能,而被广泛应用。
3.基于过盈螺烯类分子马达的手性动态可调控的胆甾相液晶,反射带隙可调,且因为分子马达对紫外光的大量的吸收可形成在螺距方向的光辐照强度的梯度,进而形成梯度的螺距,实现宽波。将此类丙烯酸酯类的分子马达的衍生物和可聚合的液晶单体结合,可制备机器人抓手、高分子振荡器等智能软体材料。所以此分子在智能软体机器人、彩色光子晶体或者宽波节能智能窗等领域都有着广泛的研究和重要的应用。
4.目前,手性过盈螺烯类分子马达的应用中,分子主体或者各类衍生物只能够对光源刺激响应,只具有单一的响应性能。然而,具有多重响应性的手性过盈螺烯类分子马达的合成与制备将具有更加广泛的应用,但是鲜有人研究。
技术实现要素:
5.基于此,本发明提供了一种具有光热双重响应性的手性过盈螺烯类分子马达,以解决现有手性过盈螺烯类分子马达只具有单一的响应性能的技术问题。
6.为实现上述目的,本发明提供了一种具有光热双重响应性的手性过盈螺烯类分子马达,该具有光热双重响应性的手性过盈螺烯类分子马达的结构通式如下式(1)所示:
[0007][0008]
其中,r
1-r8独立选自h,或者(2)-(11)式中的任一基团,且r
1-r8中至少有一个基团选自式(11),(2)-(11)式中的n为0-20之间的整数;x选自(12)-(15)式中的任一基团;y为0或者1;m为0或者1。
[0009]
根据本发明的另一方面,本发明该提供了一种具有光热双重响应性的手性过盈螺烯类分子马达的制备方法,其特征在于,包括以下步骤:
[0010]
将硫酮类化合物和叠氮化合物在催化剂的作用下发生偶联反应生成带有活性位点的光驱动手性可翻转的过盈螺烯类化合物;
[0011]
将过盈螺烯类化合物与胆固醇或其衍生物反应以在过盈螺烯类化合物的至少一个活性位点接枝胆固醇基团;
[0012]
作为本发明的进一步优选技术方案,所述叠氮化合物具有如下任意一种的结构:
[0013][0014]
其中r
1-r8独立选自h,或者(2)-(11)式中的基团,x选自(12)-(15)式中的任一基
团;
[0015]
作为本发明的进一步优选技术方案,所述硫酮化合物具有如下任意一种的结构:
[0016][0017]
其中r
1-r8独立选自h,或者(2)-(11)式中的基团,x选自(12)-(15)式中的任一基团。
[0018]
作为本发明的进一步优选技术方案,所述胆固醇的衍生物具有如下的结构:
[0019][0020]
作为本发明的进一步优选技术方案,硫酮类化合物和叠氮化合物的反应中,叠氮化合物的反应当量为硫酮类化合物的1-1.5;活性位点接枝胆固醇基团的反应中,过盈螺烯类化合物的溶液中加入1-1.2当量胆固醇衍生物。
[0021]
作为本发明的进一步优选技术方案,硫酮化合物通过酮类化合物与劳森试剂反应得到。
[0022]
作为本发明的进一步优选技术方案,酮类化合物通过萘或者萘的衍生物和丙烯酸或者丙烯酸的衍生物在多聚磷酸的作用下反应生成,或者为芴酮或芴酮的衍生物。
[0023]
作为本发明的进一步优选技术方案,叠氮化合物由酮类化合物与水合肼反应后再与二氧化锰低温氧化反应得到。
[0024]
本发明的具有光热双重响应性的手性过盈螺烯类分子马达,能够在紫外光辐照下快速发生手性翻转,撤去紫外光源后,可自发热回复到初始的状态,且热回复的速率和温度呈现一级动力学相关关系;通过将胆固醇及其衍生物引入到分子马达取代基上,胆固醇基团可随光致异构化手性翻转而发生变化,使其手性大小可随温度发生变化,变化灵敏。基于此,使得本发明的手性过盈螺烯类分子马达可对多重刺激源响应,从而拓宽了其实际应用的范围。
[0025]
本发明的具有光热双重响应性的手性过盈螺烯类分子马达的制备方法,提出了一种操作简易的方法,将具有热响应性能的胆固醇衍生物和光驱动手性可翻转的过盈螺烯类化合物相结合,制备出了既能够对光响应发生光致手性翻转,又能够对热响应发生手性大小的变化的双功能分子马达;该分子马达具有胆固醇的热响应性,使得整个分子能对热刺激源响应。因此,该分子有望制备出较新一代的多响应材料,在智能软体材料,或者响应性光子晶体上有广泛的应用的前景。
附图说明
[0026]
下面结合附图和具体实施方式对本发明作进一步详细的说明。
[0027]
图1为本发明实施例1的手性过盈螺烯类分子马达在紫外光辐照前后的吸收光谱
曲线;
[0028]
图2为本发明实施例1的手性过盈螺烯类分子马达在紫外光辐照前后的圆二色光谱曲线;
[0029]
图3为本发明实施例1的手性过盈螺烯类分子马达的手性螺旋扭曲力随温度发生变化的趋势图。
[0030]
本发明目的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
[0031]
以下结合附图对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
[0032]
除有定义外,以下实施例中所用的技术术语具有与本发明创造所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
[0033]
本发明提供了一种具有光热双重响应性的手性过盈螺烯类分子马达,该化合物的结构通式如下式(1)所示:
[0034]
[0035][0036]
其中,r
1-r8独立选自h,或者(2)-(11)式中的任一基团,且r
1-r8中至少有一个基团选自式(11),(2)-(11)式中的n为0-20之间的整数;x选自(12)-(15)式中的任一基团;y为0或者1。
[0037]
该化合物在紫外光的辐照下发生光致异构化的过程中,分子的手性大小发生变化,最终发生翻转,在停止激发后可自发地回复到初始的热稳定状态。同时分子具有热响应性的胆甾醇取代基,胆甾醇分子的手性大小可随温度发生显著的变化,因此,胆甾醇接枝的分子马达的手性大小不仅可被紫外光辐照调节,甚至发生手性的翻转,同时其手性也可随温度发生变化。该化合物具有迅速的光响应性和温度依赖的热回复行为,同时也具有温度依赖的手性大小,取代基的存在不影响分子的光之异构化行为,具有广泛的应用前景。
[0038]
上述具有光热双重响应性的手性过盈螺烯类分子马达的制备方法包括以下步骤:
[0039]
按叠氮化合物的反应当量为硫酮类化合物的1-1.5,将硫酮类化合物和叠氮化合物在催化剂的作用下发生偶联反应生成带有活性位点的光驱动手性可翻转的过盈螺烯类化合物;然后将过盈螺烯类化合物的溶液中加入了1-1.2当量胆固醇或其衍生物,通过酯化和/或者醚化反应,以在过盈螺烯类化合物的活性位点接枝胆固醇基团。
[0040]
所述叠氮化合物具有如下任意一种的结构:
[0041][0042]
其中r
1-r8独立选自h,或者(2)-(11)式中的基团,x选自(12)-(15)式中的任一基团。
[0043]
所述硫酮化合物具有如下任意一种的结构:
[0044][0045]
其中r
1-r8独立选自h,或者(2)-(11)式中的基团,x选自(12)-(15)式中的任一基团。
[0046]
所述胆固醇的衍生物具有如下的结构:
[0047][0048]
具体地,硫酮化合物和叠氮类化合物发生偶联反应,生成带有活性位点的光驱动手性可翻转的过盈螺烯类化合物的具体方法如下:
[0049]
将硫酮化合物溶解在甲苯溶液中,加入1-1.2当量的叠氮化合物,室温搅拌半个小时,加入三苯基膦,室温搅拌0.5h,之后加热到90摄氏度反应36h至反应物反应完全或者全部分解,旋蒸,再将浓缩物溶解于乙醚溶剂中室温下加入碘甲烷反应24h,除去多余的三苯基膦,反应结束后,抽滤除去不溶的固体,减压蒸馏除去溶剂,柱层析进行提纯,得到带有活性位点的光驱动手性可翻转的过盈螺烯类化合物。
[0050]
具体地,胆固醇的接枝可以直接由胆固醇衍生物和带有活性位点的光驱动手性可翻转的过盈螺烯类化合物在催化剂的作用下反应得到,具体方法如下:
[0051]
将带有活性位点的手性过盈螺烯类分子马达溶解于二氯甲烷溶液中,加入二环己基碳二亚胺、4-二甲氨基吡啶等催化剂,室温磁力搅拌,再加入1-1.5当量的胆固醇衍生物,室温反应至反应结束,冷冻除去不溶物,减压旋蒸,对浓缩物进行柱层析提纯。
[0052]
在一实例中,本发明中的硫酮化合物可以通过酮类化合物与劳森试剂反应得到,其具体实施如下:
[0053]
将酮类化合物溶解在甲苯溶剂中,加热至90℃磁力搅拌,随后加入1-1.2当量的劳森试剂,反应的过程中反应的溶剂逐渐变为蓝色或者紫色,反应10h至反应结束,减压旋蒸,对浓缩物用柱层析的方法进行提纯,得到硫酮化合物。需要注意的是,硫酮化合物极其不稳定,极容易分解,需要马上投入到下一步的反应当中。
[0054]
在另一实例中,本发明中的酮类化合物还可以通过萘或者萘的衍生物和丙烯酸或者丙烯酸的衍生物在多聚磷酸的作用下反应生成,或者酮类化合物直接选取为芴酮或芴酮的衍生物。其中萘或其衍生物与丙烯酸或其衍生物在多聚磷酸中的反应的具体实施如下:
[0055]
将萘或其衍生物和多聚磷酸混合,置于反应容器当中,加热至50℃机械搅拌,并搅拌一段时间,陆续加入3-6当量的丙烯酸或其衍生物,升温加热至70摄氏度搅拌5h,反应结束后,通过用饱和碳酸氢钠溶液、饱和食盐水、去离子水等多次重复萃取,再用柱层析硅胶法对产物进行提纯,最后的产物呈现淡黄色固体粉末或者白色固体粉末。
[0056]
在一实例中,本发明中的叠氮化合物由酮类化合物与水合肼反应后再与二氧化锰低温氧化反应得到,其具体实施如下:
[0057]
将酮类化合物溶解于一定量的乙醇当中,加热至80℃回流,磁力搅拌,再加入1-1.5当量的水合肼,反应结束后将反应物冷却至室温,再降温,使用重结晶的方法对产物进行提纯,反应物呈现为黄色的针状晶体或者为黄色的油状液体;将此反应物加入到二氯甲烷溶液当中,反应温度应该控制在-5-0℃期间,加入5当量的二氧化锰粉末,反应7h直至反应结束,真空减压抽滤,取滤液,柱层析提纯产物。叠氮类化合物在温度高时容易分解并产生副产物,所以其应该保存在-20℃左右的冷藏环境中。
[0058]
本发明提供的具有光热双重响应性的手性过盈螺烯类分子马达的潜在应用包括但不限于:
[0059]
1)基于分子的手性的大小变化和翻转的特性,能够在胆甾相液晶的反射带隙的变化、光致结构色变化、宽波反射、光写入液晶显示领域的应用;
[0060]
2)基于分子马达的光热响应特性,将其与可聚合的液晶单体结合,制备出彩色光子晶体薄膜,用于彩色光子标签、防伪标签等领域
[0061]
3)基于分子的光致异构化特性的胆甾相涂层的制备,用光或者热刺激源实现分子马达的翻转,进而带动微玻璃棒等物体的旋转运动。
[0062]
为了让本领域的技术人员更好地理解并实现本发明的技术方案,以下将通过具体实施例对本发明的技术方案作进一步地详细说明。
[0063]
实施例1:
[0064]
具有光热双重响应性的手性过盈螺烯类分子马达,以r8结构为胆固醇基团,r
1-r7结构均为h,x为甲基,m为0,y为0,其分子结构具体如下所示:
[0065][0066]
该化合物的合成方法如下:
[0067]
步骤1、将10g一甲氧基萘溶于200ml多聚磷酸置于烧瓶内,升温至50℃并机械搅拌,陆续加入10g的甲基丙烯酸,机械搅拌反应8h,然后将反应液用冰块降至室温后,用二氯甲烷萃取,再分别用饱和碳酸氢钠水溶液和去离子水洗涤两次,分液除去水层,再用无水硫酸镁吸收残留的水份,减压蒸馏除去溶剂后过柱提纯,淋洗剂为乙酸乙酯和石油醚1:5的混合溶剂,减压旋蒸除去溶剂得到白色粉末7.2g,产率67%。
[0068]
步骤2、将步骤1中的产物(5g,22.095mol)溶解于甲苯溶剂当中,加热到90℃,加入alcl3(4.4g,22mmol),持续反应五个小时,向反应物中加入水,用乙酸乙酯萃取,有机层用0.5m的盐酸溶液和水溶液洗涤三次,之后溶液用无水硫酸镁干燥。旋蒸除去溶剂,粗产物用柱层析硅胶法进行提纯,石油醚:乙酸乙酯=2:1,生成橘黄热固体(1.2g,51%产率)。
[0069]
步骤3、将步骤2的产物(2g,9.45mmol)和k2co3(1.46g,10.5mmol)溶解于n,n二甲基甲酰胺当中,加热到50℃后加入氯乙酸叔丁酯,然后升温到80摄氏度反应5个小时,反应
液冷却到室温,随后旋蒸,提取粗产物,并用柱层析法进行提纯,石油醚:乙酸乙酯比例为5:2,产物为黄色固体(3.1g,82%)。
[0070]
步骤4、将步骤3的产物(50mg,0.15mmol)溶解于四氢呋喃溶剂当中,加入劳森试剂,反应物加热到50摄氏度反应5h,旋蒸,取粗产物,用柱层析硅胶法进行提纯,石油醚:乙酸乙酯=10:1,产物为紫红色固体(40mg,78%)。
[0071]
步骤5、将步骤4的产物(540mg,1.58mmol)溶解于甲苯溶液当中,再加入叠氮化合物和三苯基膦,溶剂在室温下搅拌3h,之后升温到回流反应13h,反应中间体减压旋蒸然后重新溶解于乙醚溶剂当中(25ml),加入碘甲烷(0.3ml)并且在室温下搅拌12h,产生白色的沉淀物,对反应物抽滤旋蒸浓缩,对粗产物进行柱层析提纯,淋洗剂比例为石油醚:乙酸乙酯=2:1,产物为黄色固体,产率为52%。
[0072]
步骤6、将步骤5的产物(1g,2mmol)溶解于四氢呋喃溶剂当中,将反应装置放置在冰水浴中,缓慢加入lialh4(700mg,18.4mmol),在0摄氏度下搅拌5h,反应结束后反应液用过量的na2so4.10h2o淬灭。之后将反应物放置于室温下,抽滤除去固体,并用乙酸乙酯溶剂反复冲洗几遍,取滤液减压旋蒸,所得浓缩物用柱层析法进行提纯,淋洗剂比例为石油醚:乙酸乙酯=2:1,产物为黄色固体,产率为58.8%。
[0073]
步骤7、将步骤6的产物(0.5g,1.24mmol),以及吡啶(0.22ml,2.5mmol)、二甲基氨基吡啶(14mg)和氯甲酸胆固醇(0.6g,1.36mmol)溶解在干燥的二氯甲烷溶液当中,磁力搅拌,反应 48h,反应结束后,反应液用饱和食盐水和去离子水清洗三遍,最后用无水硫酸镁干燥,减压抽滤蒸馏取浓缩物,对粗产物用柱层析方法进行提纯,淋洗剂的比例为石油醚:乙酸乙酯=5: 1,终产物为黄色固体,产率为54%。
[0074]
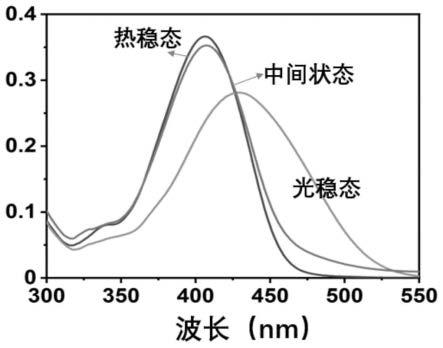
对步骤7的得到的产物进行测试,测试其紫外吸收光谱和圆二色谱在紫外光辐照前后的变化,如图1、图2所示,以表征其光响应性能和手性翻转;同时使用偏光显微镜测试分子在斜劈盒中螺距随温度的变化规律,如图3所示,随着温度的升高,条纹间距逐渐变小,对应的螺旋扭曲力变大,表现出手性随温度的变化。
[0075]
实施例2:
[0076]
具有光热双重响应性的手性过盈螺烯类分子马达,以r1、r6、r8结构为胆固醇甲酰氯基团,其余基团结构均为h,x为甲基,m为0,y为0,具体分子结构如下所示:
[0077][0078]
该化合物的合成方法如下:
[0079]
步骤1、将二羟基芴酮(10g,)和k2co3(5.3g)溶解于n,n二甲基甲酰胺当中,加热到 50℃后加入氯乙酸叔丁酯(8g),然后升温到80℃反应8个小时,反应液冷却到室温,抽滤除去固体不溶解的盐类物质,对滤液旋蒸提取粗产物,用乙醇进行重结晶,得到产物为黄色固体(8g,产率为82%)。
[0080]
步骤2、将步骤1的产物溶于300ml水合肼与50ml乙醇的混合溶剂,加入0.5ml醋酸,搅拌反应8h,将重结晶出的固体溶于二氯甲烷,加入7.06g二氧化锰,在冰水浴中搅拌8h,得到的产物过滤除去固体,滤液减压蒸馏除去溶剂并过柱提纯,淋洗剂为石油醚和三乙胺20:1 的混合溶剂,干燥后得到暖橙色固体3g,产率52%;产物放入-20℃冰箱内冷藏。
[0081]
步骤3、将8g一甲氧基萘溶于150ml多聚磷酸置于烧瓶内,升温至80℃并机械搅拌,陆续加入6g的甲基丙烯酸,机械搅拌反应5h,然后将反应液用冰块降至室温后,用二氯甲烷萃取,再分别用饱和碳酸氢钠水溶液和去离子水洗涤两次,分液除去水层,再用无水硫酸镁吸收残留的水份,减压蒸馏除去溶剂后,柱层析提纯,淋洗剂为乙酸乙酯和石油醚1:5的混合溶剂,减压旋蒸,除去溶剂得到白色粉末5.6g,产率67%。
[0082]
步骤4、将步骤3中的产物(1g)溶解于甲苯溶剂当中,加热到90℃,加入alcl3(2g),持续反应五个小时,向反应物中加入水,用乙酸乙酯萃取,有机层用0.5m的盐酸溶液和水溶液洗涤三次,之后溶液用无水硫酸镁干燥。旋蒸除去溶剂,粗产物用柱层析硅胶法进行提纯,石油醚:乙酸乙酯=2:1,生成橘黄热固体(0.5g,产率50%)。
[0083]
步骤5、将步骤4的产物(2g,9.45mmol)和k2co3(1.46g,10.5mmol)溶解于n,n二甲基甲酰胺当中,加热到50℃后加入氯乙酸叔丁酯,然后升温到80摄氏度反应5个小时,反应液冷却到室温,旋蒸提取粗产物,并用柱层析法进行提纯,石油醚:乙酸乙酯比例为5:2,产物为黄色固体(3.1g,82%)。
[0084]
步骤6、将步骤5的产物(1g)溶解于四氢呋喃溶剂当中,加入1.2当量的劳森试剂,反应物加热到60摄氏度反应4.5h,旋蒸取粗产物,用柱层析硅胶法进行提纯,淋洗剂比例为石油醚:乙酸乙酯=10:1,产物为紫红色固体(60mg,78%)。
[0085]
步骤7、将步骤2的产物1.64g和步骤6的产物1.89g,溶解在100ml甲苯溶液当中,在
室温下搅拌一个小时之后加入三苯基膦,再在室温下反应3h,之后油浴加热升温至90℃,直至反应结束8h,过滤除去固体,滤液减压蒸馏除去溶剂后过柱提纯,淋洗剂为乙酸乙酯和石油醚5:1的混合溶剂,干燥后得到黄色固体0.5g,产率31.2%。
[0086]
步骤8、将步骤7的产物(0.5g)溶解于四氢呋喃溶剂当中,反应温度控制为0摄氏度,缓慢加入九倍当量的lialh4(2g),控制在冰水浴中搅拌5h,反应结束后反应液用过量的 na2so4.10h2o淬灭;之后将反应物放置于室温下,抽滤除去固体,并用乙酸乙酯溶剂反复冲洗几遍,滤液减压旋蒸的浓缩物,用柱层析法进行提纯,淋洗剂比例为甲醇:乙酸乙酯=1: 20,产物为黄色固体,产率为78.8%。
[0087]
步骤9、将步骤8的产物(0.5g)、二环己基碳二亚胺(0.3g)、二甲基氨基吡啶(14mg)和胆固醇甲酸(0.75g)溶解在干燥的二氯甲烷溶液当中,磁力搅拌,反应36h,反应结束后,反应液减压抽滤蒸馏取浓缩物,对粗产物用柱层析方法进行提纯,淋洗剂的比例为石油醚:乙酸乙酯=5:1,终产物为黄色固体,产率为20.1%。
[0088]
由以上步骤可知,采用在分子马达的转子和定子部分接入相同取代基的方式,可以使用同类型的反应,采用一步合成的方法在三个活性位点接入胆固醇取代基,合成较为方便。对步骤9的产物进行表征,观察其紫外吸收光谱和圆二色谱在紫外光辐照前后的变化,可观察到类似于实施例1一样的光谱的变化,吸收光谱的红移和吸收强度的略微下降,圆二色谱的峰的红移和峰正负方向的切换,证明了分子的手性发生了翻转;同时使用偏光显微镜观察斜劈盒中条纹间距随温度的变化,具有明显的条纹间距随温度的升高而降低的实验现象,证明了该分子同样具有优异的热响应性能。
[0089]
虽然以上描述了本发明的具体实施方式,但是本领域熟练技术人员应当理解,这些仅是举例说明,可以对本实施方式做出多种变更或修改,而不背离本发明的原理和实质,本发明的保护范围仅由所附权利要求书限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1