一种JAK2激酶选择性抑制剂及其制备方法和用途与流程
一种jak2激酶选择性抑制剂及其制备方法和用途
技术领域
1.本发明涉及药物化学领域,具体地涉及一种jak2激酶选择性抑制剂及其制备方法、药物组合物和用途。
背景技术:
2.蛋白激酶家族已经成为多种疾病的主要致病因子,其中jak家族细胞蛋白酪氨酸激酶(包含jak1,jak2,jak3和tyk2)在细胞因子信号传导中起关键作用。细胞因子在结合其受体时活化jak,然后使细胞因子受体磷酸化,激活信号转导物和转录激活物(stat)家族。近年来,jak抑制剂的治疗潜能集中在影响各种免疫系统病理学情况的疾病。包括特应性、细胞介导超敏反应(过敏性接触性皮炎,过敏性肺炎)、系统性红斑狼疮(sle)、类风湿性关节炎、牛皮癣、移植(移植物排斥,移植物抗宿主病)等。芦可替尼、托法替尼等jak激酶抑制剂已经上市,用于治疗骨髓纤维化、类风湿性关节炎等疾病。
[0003][0004]
最近以来,发现了epo-jak2信号传导途经在骨髓组织增殖病症和增生型糖尿病视网膜病变中就要重要的作用。fedratinib作为一款选择性的jak2激酶抑制剂,已经获得美国fda批准上市,用于骨髓纤维化的治疗。另外,epo-jak2激酶信号通路为增生型糖尿病视网膜病变的有效生成血管因子,即影响工作年龄段的糖尿病患者视力丧失的主要原因。因此,jak2激酶抑制剂可能是治疗视网膜病变的潜在治疗药物。因此,由于可利用治疗上述jak信号传导途径失调或得到直接或间接补充的疾病的疗法不足,所以对研发用作激酶,特别是jak2的选择性激酶抑制剂的化合物存在需求。
[0005]
目前上市的jak2选择性激酶抑制剂fedratinib存在多种不足,例如针对jak2的选择性偏低,化合物生物利用度低以及毒性较高,导致临床上的安全用药受到限制。因此,需要开发新的能够克服以上缺点的jak2激酶高选择性抑制剂。
技术实现要素:
[0006]
为了克服现有技术的不足,一方面,本发明提供了一种具有下列式(i)的化合物,
[0007][0008]
或其互变异构体、同位素标记物、溶剂化物、前药以及药学上可接受的盐。
[0009]
在一个实施方案中,本发明所提供的化合物包括:
[0010]010][0011]
或其互变异构体、同位素标记物、溶剂化物、前药以及药学上可接受的盐。
[0012]
另一方面,本发明提供了一种药物组合物,其包含根据本发明所述的化合物或其互变异构体、同位素标记物、溶剂化物、前药、药学上可接受的盐,以及药学上可接受的载体。
[0013]
在一个实施方案中,本发明所提供的药物组合物任选地进一步包含一种或多种其它治疗剂,其中所述其它治疗剂选自jak激酶抑制剂或磷酸二酯酶4抑制剂。
[0014]
再一方面,本发明提供了本文所述的化合物或其互变异构体、同位素标记物、溶剂化物、前药、药学上可接受的盐以及本文所述的药物组合物在制备用于预防和/或治疗由jak2激酶介导的疾病的药物中的用途。
[0015]
在一个实施方案中,本发明所述的疾病包括但不限于骨髓组织增殖病症、炎性疾病、实体瘤或其他类型的癌症、i型糖尿病、ii型糖尿病、糖尿病并发症、哮喘、过敏反应、成
人呼吸窘迫综合征、心血管疾病、肝病、眼病、自身免疫疾病、重症肌无力、移植物抗宿主病、神经变性疾病、病毒性疾病。
[0016]
在一个实施方案中,本发明所述的疾病包括但不限于类风湿性关节炎、急性胰腺炎、慢性胰腺炎、鼻炎、特应性皮炎、溃疡性结肠炎、肾小球肾炎、慢性甲状腺炎、慢性活动性肝炎、炎性肠病、青少年关节炎、多肌炎、强直性脊柱炎、牛皮癣性关节炎、皮肤炎症、过敏性接触性皮炎、过敏性肺炎或过敏性鼻炎。
[0017]
在一个实施方案中,本发明所述的疾病包括但不限于银屑病、多发性硬化、系统性红斑狼疮(sle)、自身免疫性甲状腺病、硬皮病、格雷夫斯病、自身免疫性胃炎、自体免疫溶血性疾病、自身免疫性嗜中性白血球减少症、舍格仑综合征、因实体瘤诱导的免疫抑制。
[0018]
在一个实施方案中,本发明所述的疾病包括但不限于运动神经元疾病、阿尔茨海默病、帕金森病、肌萎缩性侧索硬化、亨廷顿舞蹈病、脑缺血症或因跌打损伤导致的神经变性疾病、中风、谷氨酸神经中毒或低氧。
[0019]
在一个实施方案中,本发明所述的疾病包括但不限于eb病毒、乙型肝炎病毒、丙型肝炎病毒、hiv、htlv1、水痘-带状疱疹病毒和/或人乳头瘤病毒引起的疾病。
[0020]
在一个实施方案中,本发明所述的疾病包括但不限于消化/胃癌、直肠癌、结肠癌、结直肠癌、肝癌、皮肤癌、胰腺癌、乳腺癌、卵巢癌、前列腺癌、淋巴瘤、白血病、肾癌、喉癌、肺癌、肌肉癌、骨癌、膀胱癌、宫颈癌、脑癌、婴儿血管瘤、黑素瘤、卡波西肉瘤、多发性骨髓瘤。
[0021]
在一个实施方案中,本发明所述的疾病选自急性髓性白血病或慢性髓性白血病(cml)。
[0022]
在一个实施方案中,本发明所述的慢性髓性白血病抵抗目前的治疗。
[0023]
在一个实施方案中,本发明所述的疾病选自与血管生成相关的病症、中风中的缺血性/再灌注损伤、心肌缺血、肾缺血、心脏病发作、心脏肥大、动脉粥样硬化、血小板减少症、器官缺氧和血小板聚集。
[0024]
在一个实施方案中,本发明所述的疾病选自眼新生血管形成、血管增生。
[0025]
在一个实施方案中,本发明所述的疾病选自真性红细胞增多症、特发性血小板增多、骨髓纤维化伴骨髓化增生。
[0026]
在一个实施方案中,本发明所述的骨髓组织增殖病症因jak家族激酶途径功能的获得而产生。
[0027]
在一个实施方案中,本发明所述的骨髓组织增殖病症作为因jak家族激酶途径功能的获得导致基因或蛋白质融合的结果而产生。
[0028]
在一个实施方案中,本发明所述的疾病选自增生性视网膜病变(包括但不限于增生性糖尿病视网膜病变)、过敏性结膜炎、特应性角结膜炎。
[0029]
在一个实施方案中,本发明所述的疾病为jak2激酶异常导致的嗜酸性粒细胞增多引起的相关疾病。
[0030]
在一个实施方案中,本发明所述的疾病包括重度嗜酸粒细胞性哮喘(sea)、嗜酸性肉芽肿性多血管炎(egpa)、高嗜酸粒细胞综合征(hes)和慢性鼻-鼻窦炎伴鼻息肉(crswnp)。
[0031]
在一个实施方案中,本发明提供的药物组合物可以制成以下固体或液体形式的剂型,例如片剂、胶囊剂、丸剂、颗粒剂、散剂、粉剂、栓剂、贴剂、溶液剂、混悬剂、乳剂、酊剂、糖
浆剂、喷雾剂等;优选地,本发明提供的药物组合物可以制成片剂、胶囊剂、注射剂、吸入剂、滴眼液、外用制剂等;更优选地,本发明提供的药物组合物可以制成片剂和滴眼剂。
[0032]
在一个实施方案中,本发明的药物组合物被制成滴眼剂,其包含本发明化合物、增稠剂、渗透压调节剂、ph调节剂、抑菌剂及药学上可接受的其他辅料;其中,本发明化合物在滴眼剂中的重量百分比含量为0.05%~1%,优选0.1%~0.5%。
[0033]
在一个实施方案中,本发明的药物组合物被制成滴眼剂,其特征在于,所述增稠剂在滴眼剂中的重量百分比含量为0%~5%,优选0.5%~1%;所述增稠剂选自羧甲基纤维素钠、羟丙甲纤维素、聚乙烯醇、透明质酸、甘油之一或其任意组合。
[0034]
在一个实施方案中,本发明的药物组合物被制成滴眼剂,其特征在于,所述渗透压调节剂在滴眼剂中的重量百分比含量为0%~5%,优选0.1~1%;所述的渗透压调节剂选自氯化钠、葡萄糖、硼酸、硼砂之一或其任意组合;所述抗氧化剂优选为氯化钠或葡萄糖。
[0035]
在一个实施方案中,本发明的药物组合物被制成滴眼剂,其特征在于,所述ph调节剂在滴眼剂剂中的重量百分比含量为0%~10%,优选0.5%~5%;所述的保湿剂选自盐酸、氢氧化钠、碳酸钾、碳酸氢钠、硼酸、硼砂之一或其任意组合;所述ph调节剂可使滴眼剂的ph值范围落在4~9之间,优选ph值范围为5~7。
[0036]
在一个实施方案中,本发明的药物组合物被制成滴眼剂,其特征在于,所述抑菌剂在滴眼剂中的重量百分比含量为0%~1%,优选0.001%~0.5%;所述的抑菌剂选自对羟基苯甲酸乙酯、苯甲酸、山梨酸、乙醇、三氯叔丁醇、苯扎氯铵、苯扎溴铵之一或其任意组合。
[0037]
在一个实施方案中,提供了本发明所述的滴眼剂的制备方法,包括以下步骤:
[0038]
(1)采用研磨、过筛或气流粉碎方法将本发明所述化合物进行粉碎,制得原料药粉末;其中d90粒径控制在≤30μm;
[0039]
(2)将处方量的增稠剂、渗透压调节剂加入适量纯水中,在40~80℃水浴条件下充分搅拌均匀,待组分完全溶解后冷却,得到溶液;
[0040]
(3)将步骤(1)所得的原料药粉末加入步骤(2)中所得的溶液中,充分搅拌,使其完全溶解;
[0041]
(4)向步骤(3)所得的混合液中加入ph调节剂、抑菌剂等辅料,继续搅拌至完全溶解;
[0042]
(5)将步骤(4)所得的药液用纯水定容至目标体积,灌装,即得本发明所述的滴眼剂。
具体实施方式
[0043]
实施例1
[0044]
步骤1 2-氯-5-(三氟甲基)嘧啶-4-胺(2)
[0045][0046]
向100ml单口瓶中依次加入化合物1(2.00g,9.21mmol)、四氢呋喃(20ml)和27%wt氨水(2ml),室温反应过夜。tlc(v
石油醚
:v
乙酸乙酯
=5:1)监测原料反应完全,加入乙酸乙酯
(100ml)稀释,饱和氯化钠溶液(50ml
×
3)洗涤,无水硫酸钠干燥,浓缩,柱层析纯化(v
石油醚
:v
乙酸乙酯
=10:1),得到500mg白色固体2,收率27.5%。1h nmr(600mhz,cd3od)δ(ppm):8.30(s,1h,arh)
[0047]
步骤2 n-叔丁基-3-((2-氯-5-(三氟甲基)嘧啶-4-基)氨基)苯磺酰胺(3)
[0048][0049]
向100ml单口瓶中依次加入化合物2(1.00g,5.06mmol)、1,4-二氧六环(50ml)、碳酸铯(3.33g,10.1mmol)、n-叔丁基-3-溴苯磺酰胺(1.77g,6.07mmol)、pd2dba3(460mg,0.51mmol)和xantphos(590mg,1.00mmol),氮气保护,100℃反应过夜。tlc(v
石油醚
:v
乙酸乙酯
=5:1)监测原料反应完全,冷却至室温,加水(60ml)淬灭反应,乙酸乙酯(50ml
×
3)萃取,合并有机相,饱和氯化钠溶液(100ml)洗涤,无水硫酸钠干燥,浓缩,柱层析纯化(v
石油醚
:v
乙酸乙酯
=4:1),得到1.07g白色固体3,收率51.7%。1h nmr(600mhz,dmso-d6)δ(ppm):9.73(s,1h,nh),8.64(s,1h,nh),7.92(t,j=1.8hz,1h,arh),7.71-7.67(m,1h,arh),7.67-7.64(m,1h,arh),7.59(t,j=7.8hz,1h,arh),7.55(s,1h,arh),1.13(s,9h,ch3).
[0050]
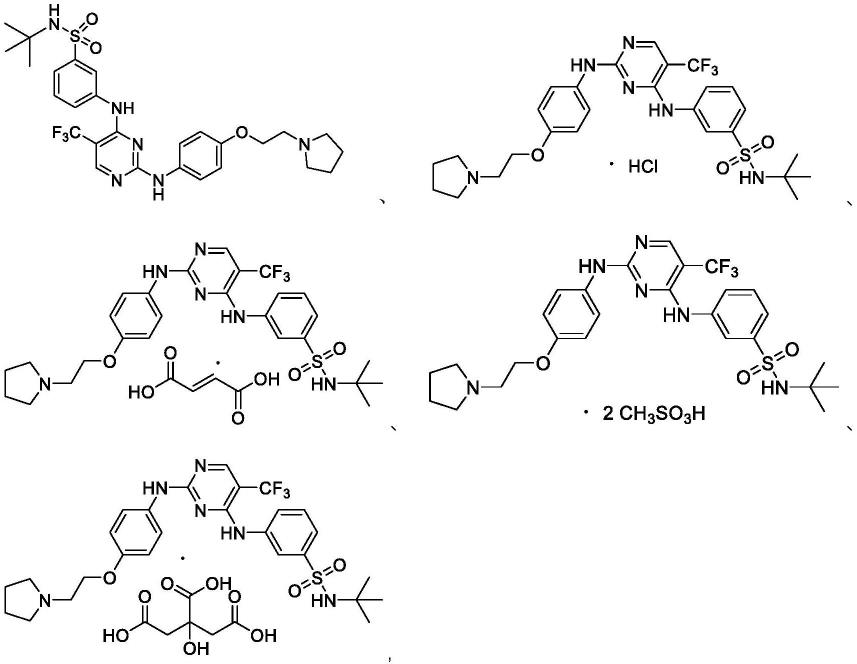
步骤3 n-叔丁基-3-((5-三氟甲基-2-(4-(2-(吡咯烷-1-基)乙氧基)苯胺基)嘧啶-4-基)氨基)苯磺酰胺(实施例1)
[0051][0052]
向50ml单口瓶中依次加入化合物3(550g,1.34mmol)、正丁醇(10ml)、对甲苯磺酸一水合物(256mg,1.34mmol)和4-[2-(吡咯烷-1-基)乙氧基]苯胺(250mg,1.21mmol),氮气保护,60℃反应2h。tlc(v
二氯甲烷
:v
甲醇
=10:1)监测原料反应完全,冷却至室温,加水(30ml)淬灭反应,乙酸乙酯(20ml
×
3)萃取,合并有机相,饱和碳酸氢钠溶液(30ml)洗涤,饱和氯化钠溶液(30ml)洗涤,无水硫酸钠干燥,浓缩,柱层析纯化(v
二氯甲烷
:v
甲醇
=100:1),再经hplc制备,得到310mg白色固体实施例1,收率39.8%。1h nmr(600mhz,dmso-d6)δ(ppm):9.35(br,1h,nh),8.95(s,1h,nh),8.25(s,1h,nh),7.87-7.55(m,3h,arh),7.54-7.22(m,4h,arh),6.68(s,2h,arh),3.96(t,j=6.0hz,2h,och2),2.72(t,j=6.0hz,2h,nch2),2.51-2.48(m,4h,四氢吡咯-ch2),1.69-1.67(m,4h,四氢吡咯-ch2),1.09(s,9h,ch3).hrms(esi):m/z[m+h]
+
理论值c
27h33
f3n6o3s:579.2365;实测值579.2340.
[0053]
实施例2 n-叔丁基-3-((5-三氟甲基-2-(4-(2-吡咯烷-1-乙氧基)苯胺基)嘧啶-4-基)氨基)苯磺酰胺盐酸盐(实施例2)
[0054][0055]
向25ml单口瓶中依次加入化合物实施例1(120mg,0.24mml)和hcl/ea溶液(6ml,3n),氮气保护,室温反应3h。tlc(v
二氯甲烷
:v
甲醇
=10:1)监测原料反应完全,过滤,滤饼溶于水(5ml)中,冻干,得到110mg白色固体实施例2,收率81.4%。1h nmr(600mhz,dmso-d6)δ(ppm):10.88(br,1h,hcl),10.06(br,1h,nh),9.35(br,1h,nh),8.43(s,1h,nh),7.83-7.71(m,3h,arh),7.61-7.53(m,2h,arh),7.35(s,2h,arh),6.79(s,2h,arh),4.32-4.26(m,2h,och2),3.58-3.52(m,4h,四氢吡咯-ch2),3.11-3.05(m,2h,nch2),2.04-1.98(m,2h,四氢吡咯-ch2),1.92-1.85(m,2h,四氢吡咯-ch2),1.09(s,9h,ch3);hrms(esi):m/z[m+h]
+
理论值c
27h33
f3n6o3s:579.2365;实测值579.2354.
[0056]
实施例3 n-叔丁基-3-((5-三氟甲基-2-(4-(2-吡咯烷-1-乙氧基)苯胺基)嘧啶-4-基)氨基)苯磺酰胺富马酸盐(实施例3)
[0057][0058]
向25ml单口瓶中依次加入实施例1(200mg,0.34mmol)、富马酸(40mg,0.34mmol)、甲醇(2ml)和水(2ml),加毕,60℃搅拌1h。减压蒸馏除去溶剂得黄色粉末,经45℃真空干燥5h,得到222mg黄色固体实施例3,收率94.1%。1h nmr(400mhz,dmso-d6)δ(ppm):9.63(br,1h,cooh),8.98(br,1h,cooh),8.37(s,1h,arh),7.86(s,1h,nh),7.77(s,1h,nh),7.67(d,j=7.8hz,1h,arh),7.61-7.51(m,2h,arh),7.50-7.46(m,1h,arh),7.41(s,2h,arh,nh),7.11(d,j=7.9hz,1h,arh),6.78(s,2h,arh),6.63(s,2h,ch=ch),4.23(s,2h,och2),3.52(t,j=4.9hz,2h,nch2),3.34(s,4h,nch2),2.01-1.89(m,4h,ch2),1.10(s,9h,c(ch3)3).
[0059]
实施例4 n-叔丁基-3-((5-三氟甲基-2-(4-(2-吡咯烷-1-乙氧基)苯胺基)嘧啶-4-基)氨基)苯磺酰胺柠檬酸盐(实施例4)
[0060][0061]
向25ml单口瓶中依次加入实施例1(200mg,0.34mmol)、柠檬酸(71mg,0.34mmol)、
甲醇(2ml)和水(2ml),加毕,60℃搅拌1h。减压蒸馏除去溶剂得黄色粉末,经45℃真空干燥5h,得到231mg黄色固体实施例4,收率88.2%。1h nmr(400mhz,dmso-d6)δ(ppm):9.63(s,1h,cooh),8.98(s,1h,cooh),8.37(s,1h,arh),7.86(s,1h,nh),7.76(s,1h,nh),7.67(d,j=7.8hz,1h,arh),7.60-7.51(m,2h,arh),7.51-7.46(m,1h,arh),7.41(s,2h,arh,nh),7.11(d,j=7.9hz,1h,arh),6.79(s,2h,arh),4.23(s,1h,och2),3.54(t,j=4.9hz,2h,nch2),3.39(s,4h,ch2),2.74(d,j=15.4hz,2h,coch2),2.64(d,j=15.4hz,2h,coch2),1.95(s,4h,ch2),1.10(s,9h,c(ch3)3).
[0062]
实施例5 n-叔丁基-3-((5-三氟甲基-2-(4-(2-吡咯烷-1-乙氧基)苯胺基)嘧啶-4-基)氨基)苯磺酰胺二甲磺酸盐(实施例5)
[0063][0064]
向25ml单口瓶中依次加入实施例1(200mg,0.34mmol)、甲磺酸(65mg,0.68mmol)、甲醇(2ml)和水(2ml),加毕,60℃搅拌1h。减压蒸馏除去溶剂得黄色油状物,经45℃真空干燥5h,得到239mg黄色固体实施例5,收率91.2%。1h nmr(400mhz,dmso-d6)δ(ppm):9.69(br,1h,so3h),9.28(br,1h,so3h),8.42(s,1h,arh),7.85(s,1h,nh),7.74(s,1h,nh),7.69(d,j=7.7hz,1h,arh),7.61-7.51(m,2h,arh),7.50-7.45(m,1h,arh),7.38(s,2h,arh,nh),7.12(d,j=7.8hz,1h,arh),6.81(s,2h,arh),4.22(t,j=4.9hz,2h,och2),3.64-3.53(m,4h,nch2),3.17-3.06(m,2h,nch2),2.37(s,6h,ch3so3),2.09-1.95(m,2h,ch2),1.95-1.82(m,2h,ch2),1.10(s,9h,c(ch3)3)
[0065]
实施例6不同ph调节剂对处方的影响
[0066]
按照表1中的处方制备滴眼剂。
[0067]
表1
[0068][0069]
制备方法如下:
[0070]
a液制备:分别取适量的纯水(约90ml),加入相应量的羟丙基纤维素、氯化钠,60℃
水浴条件下搅拌至完全溶解,冷却至室温备用。
[0071]
分别往上述制备的a液中加入相应量的实施例2化合物,充分搅拌至完全溶解。继续加入不同的ph调节剂(分别是硼酸-硼砂体系、氢氧化钠-磷酸二氢钾体系、碳酸氢钠体系),充分搅拌至完全溶解;继续加入抑菌剂(对羟基苯甲酸乙酯)等,待完全溶解后,用纯水定容至100ml,搅拌均匀即得。
[0072]
根据处方1、处方2和处方3制备得到的滴眼剂的ph值分别为5.6、5.9和6.0。处方3的样品溶液放置后逐渐出现浑浊的现象。
[0073]
实施例7不同增稠剂对处方的影响
[0074]
按照表2中的处方制备滴眼剂。
[0075]
表2
[0076][0077][0078]
制备方法如下:
[0079]
a液制备:分别取适量的纯水(约90ml),加入相应量的增稠剂(分别是羟丙基纤维素、透明质酸、甘油),分别加入相应量的氯化钠,60℃水浴条件下搅拌至完全溶解,冷却至室温备用。
[0080]
分别往上述制备的a液中加入相应量的hy-0708,充分搅拌至完全溶解。继续加入ph调节剂硼酸-硼砂体系,充分搅拌至完全溶解;继续加入抑菌剂等,待完全溶解后,用纯水定容至100ml,搅拌均匀即得。
[0081]
根据处方4、处方5和处方6制备得到的滴眼剂均为澄清透明状,黏度在分别为36mpa
·
s、47mpa
·
s和41mpa
·
s。
[0082]
生物活性测试
[0083]
(一)激酶抑制活性及选择性
[0084]
1.实验材料
[0085]
表3相关实验材料和和试剂厂家信息
[0086][0087]
2.实验步骤
[0088]
2.1配制1x激酶反应缓冲液:
[0089]
依据表4配制1x激酶反应缓冲液,进行相关实验操作
[0090]
表4配制好的1x激酶反应缓冲液
[0091][0092]
2.2激酶反应条件:
[0093]
表5激酶反应条件
[0094][0095]
2.3实验步骤
[0096]
在稀释板中用dmso对化合物进行3倍梯度稀释,化合物起始浓度为10um。将化合物50倍稀释到1x激酶反应缓冲液中,在振荡器上震荡20分钟。用1x的酶反应缓冲液配制准备2x激酶。向反应板中每孔加入2μl激酶。向每孔加入1μl在缓冲液中稀释好的化合物,用封板膜封住板子,1000g离心30秒,室温放置10分钟。用1x的酶反应缓冲液配制4xatp/底物混合
液,向反应板中加入1μl 4x atp/底物混合液。用封板膜封住板子1000g离心30秒,室温反应60分钟。转移4μl adp-glo到384反应板中,1000rpm/min离心1min,25℃孵育40min。转移8μl detection溶液到384反应板中,1000rpm/min离心1min,25℃孵育40min。使用bmg多功能读板机读取rlu(relative luminescence unit)信号。信号强度用于表征激酶的活性程度。
[0097]
3.数据分析
[0098]
化合物抑制率(%)=100%-(化合物-阳性对照)/(阴性对照-阳性对照)*100%。ic
50
通过抑制率由prism graphpad7.0计算。
[0099]
4.实验结果
[0100]
从表6可知,本发明实施例1化合物对jak2激酶具有显著的抑制活性,与fedratinib相当。但是,相对于jak1、jak3及tyk2而言,本发明实施例1化合物对jak2的选择性要显著优于fedratinib。
[0101]
表6本发明化合物的激酶抑制活性和选择性
[0102][0103]
(二)大鼠体内药代动力学参数
[0104]
1.实验材料
[0105]
实施例1化合物(纯度:99.059%),内标名称:茶碱(99.9%),sd大鼠(spf级,6-7周龄),8只,雄性(220
±
20g)。许可证:scxk(湘)2019-0004
[0106]
2.实验方法
[0107]
灌胃组:供试品研磨后,采用0.5%cmc-na助溶。静注组:5%dmso+5%聚乙二醇-12-羟基二醇(solutol)+90%生理盐水。取大鼠8只,随机分成2组、每组4只,雄性,于给药前禁食12h,自由饮水。静脉组尾静脉注射给药2ml/kg,口服组灌胃给药10mg/kg,于给药后5min,15min,30min,1h,2h,4h,6h,8h和24h时间点采集血样;血浆采集用肝素钠作为抗凝剂,采集后的血样及时安排进行离心处理(离心转速3500rpm,离心时间10min),离心得到的血浆及时转至ep管中,冷冻保存于-20℃冰箱,待测。开发lc-ms/ms定量测定血浆样品中药物浓度;并进行部分验证,部分验证的内容包括:考察提取回收率、精密度、准确度、灵敏度和线性范围。样品检测采用随行标准曲线和质控样品进行检测结果准确性的控制和判断,确保检测结果的准确可靠性。将所得的血药浓度-时间数据,用phoenix winnonlin 7软件求算药代动力学参数。用矩量法求算药代动力学参数,即曲线下面积(auc)、平均驻留时间(mrt)、清除率(cl/f)、稳态表观分布容积(vss/f)分别按下列各式估算:
[0108]
auc
tn
=∑(ci+c
i-1
)(t
i-t
i-1
)/2
[0109]
auc
∞
=auc
tn
+cm/λ
[0110]
aumc
tn
=∑(t
ici
+t
i-1ci-1
)(t
i-t
i-1
)/2
[0111]
aumc
∞
=aumc
tn
+c
tn
(tn/λ+1/λ2)
[0112]
mrt=aumc
∞
/auc
∞
[0113]
cl/f=dose/auc
[0114]vss
/f=cl*mrt
[0115]
式中c
tn
为给药后最后点tn的血药浓度,k为消除速率常数,用ln(浓度)-时间直线回归求得。药物代谢动力学参数皆采用非房室模型计算。
[0116]
3.实验结果
[0117]
表7实施例1化合物的口服药代动力学参数
[0118]
[0119]
由表7可知,实施例1化合物的大鼠口服生物利用度为13.4%,对比化合物fedratinib的大鼠生物口服生物利用度为7.24%【wo2019140953】,本发明化合物的口服生物利用度显著高于fedratinib。
[0120]
(三)本发明化合物滴眼液结膜炎实验
[0121]
1.实验目的
[0122]
通过豚鼠过敏性结膜炎模型评价本发明化合物滴眼液的抗结膜炎作用
[0123]
2.实验材料
[0124]
实验动物:hartly豚鼠40只(南京莱莱芙养殖场,合格证号:scxk(苏)2019-0005),雄性,周龄6-8周,体重300-350g;
[0125]
实验耗材:卵清蛋白(ova,阿拉丁)、豚草花粉提取物(rw,德国greer公司)、氢氧化铝、无菌生理盐水(对照、溶媒);
[0126]
受试药物:本发明实施例2化合物滴眼液低高剂量组(0.01%、0.1%)、阳性对照fedratinib剂量组(0.1%)。
[0127]
3.实验方法
[0128]
hartly豚鼠在第0、2、4、6、8天腹腔注射含ova 100μg和al(oh)
3 5mg的pbs液200μl,在第10、11、12、13、14天将ova(每眼750μg)溶于20μlpbs点眼激发。每组6只,共6组。第10天开始给药,每天2次,连续给药5天,在给药后1h用裂隙灯进行评分。
[0129]
4.指标测定
[0130]
临床评分:各组豚鼠于实验第10、11、12、13、14天,ova过敏液激发液或生理盐水点眼后1h在裂隙灯下对眼表炎症进行严重程度的评估,并拍照记录。包括眼睑水肿、结膜水肿、结膜充血、溢泪在内的四个方面进行评分,根据其各项表现的严重程度评分0-3分不等。四项指标的总分在0-12分之间。
[0131]
表8.过敏性结膜炎评分标准
[0132][0133][0134]
5.实验结果
[0135]
表9.本发明滴眼液治疗豚鼠过敏性结膜炎评分
[0136][0137]
#
p《0.05,
##
p《0.01vs模型;*p《0.05,**p《0.01vs fedratinib
[0138]
实验结果显示:与模型组相比,实施例2滴眼液低高剂量组(0.01%、0.1%)、fedratinib组(0.1%)均能显著性改善豚鼠过敏性结膜炎的评分(p《0.05,p《0.01),提示各给药组均有一定的治疗效果。令人惊奇地发现,本发明化合物即使在剂量降低10倍的情况下,其仍然表现出优于fedratinib的治疗效果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1