一种电致变色聚合物及其制备方法和应用与流程
1.本发明属于电致变色材料技术领域,具体涉及一种电致变色聚合物及其制备方法和应用。
背景技术:
2.电致变色是指在外加电压的驱动下,材料的光学性能(透射率或反射率等)在可见光范围内产生稳定的可逆变化的现象。在外观上,电致变色(ec,electro chromic)材料通常表现为颜色及透明度的可逆变化。目前已有多种应用电致变色(ec)材料的电致变色(ec)装置面市,例如:用于减弱眩光的汽车后视镜,根据光线变化调节反射或吸收阳光的智能窗等。
3.在一般的电致变色混合材料中,包含n型材料和p型材料,紫精是常见的n型(即阳离子型,得到电子而变色)电致变色材料,与p型(即阴离子型,失去电子而变色)电致变色材料在溶液中形成互补写-擦(writing-erasing)体系。在电场作用下,体系中两种互补离子可自由扩散或迁移到电解质表层,紫精从电极处获得电子,由氧化态转变到还原态,对应的阴离子材料在电极上失去电子,由还原态转变为氧化态,材料由无色变为有色;自擦除(self-erasing)过程中,两种离子从电解质表层迁移到溶液内部,相遇后电子转移,材料褪色,常见的p型材料如吩嗪、吩噻衍生物等。
4.聚(3,4-丙烯二氧基噻吩)作为电致变色材料具有光学对比度大,响应速度快的优点,具有极大的工业化的应用潜力。然而,能否在实际中应用,电致变色器件的稳定性起着决定性的作用。对其稳定性影响起着重要作用的分别是其组成的三部分:电致变色层,电解质层以及离子存储层。关于电致变色的光学性能衰减的机理,有研究认为是氧化还原的循环过程中,离子的掺杂与脱掺杂的不完全导致活性位点被覆盖,引起了电致变色的衰减。所以提出选择与电致变色材料适配的掺杂离子可以有效地改善电致变色的稳定性。电致变色的器件在工作时还涉及到了离子的迁移传输。离子存储层与电致变色层的电荷平衡至关重要。选用聚(3,4-丙烯二氧吡咯)(mccp)作为pprodot的对电极材料,形成电荷量的平衡,有效地提升了器件的稳定性。然而,电致变色器件最重要的部分是电致变色层,上述的适配可能具有唯一性。所以从电致变色材料本身结构的角度来提升器件的稳定性至关重要。因此,如何提供一种具有较大溶解度,且不会产生较大的空间位阻的嵌段聚合物,使其作为电致变色材料制备电致变色层,已成为目前亟待解决的技术问题。
技术实现要素:
5.针对现有技术的不足,本发明的目的在于提供一种电致变色聚合物及其制备方法和应用。本发明中,通过对电致变色聚合物的结构进行设计,制备的电致变色聚合物具有较大的溶解度,且不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入,适用于作为电致变色材料,制备电致变色装置。
6.为达此目的,本发明采用以下技术方案:
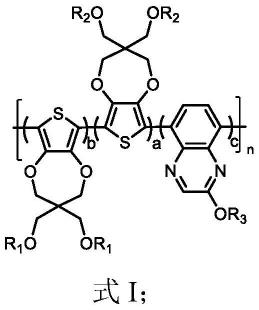
7.第一方面,本发明提供一种电致变色聚合物,所述电致变色聚合物具有如式i所示结构:
[0008][0009]
其中,r1和r2各自独立地选自氢原子、c2~c20酰基、c1~c20直链或支链烷基中的任意一种,r3为c1~c18直链或支链烷基;
[0010]
a、c为大于0的整数,b为≥0的整数;
[0011]
n为3~100的整数。
[0012]
本发明中,通过对电致变色聚合物的结构进行设计,使电致变色聚合物具有较大的平面性,可以形成致密的分子堆积,且喹喔啉基团上具有的两个n原子具有较强的吸电子能力,通过在喹喔啉基团上引入单个烷氧基,可以有效的提升电致变色聚合物的溶解性,且制备得到的电致变色聚合物不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入。本发明提供的电致变色聚合物适用于作为电致变色材料,制备电致变色装置。
[0013]
本发明中,所述c2~c20可以是c2、c4、c6、c8、c10、c12、c14、c16、c18或c20等。
[0014]
所述c1~c20可以是c1、c2、c4、c6、c8、c10、c12、c14、c16、c18或c20等。
[0015]
所述c1~c18可以是c1、c2、c4、c6、c8、c10、c12、c14、c16或c18等。
[0016]
n可以是3、5、10、20、30、40、50、60、70、80、90或100等。
[0017]
以下作为本发明的优选技术方案,但不作为对本发明提供的技术方案的限制,通过以下优选的技术方案,可以更好的达到和实现本发明的目的和有益效果。
[0018]
作为本发明的优选技术方案,所述r1和r2各自独立地选自丁酰基、己酰基、辛酰基、2-乙基己酰基、丙基、丁基、戊基、己基、辛基或十二烷基中的任意一种。
[0019]
优选地,所述r3选自甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、十二烷基或十三烷基中的任意一种。
[0020]
作为本发明的优选技术方案,所述电致变色聚合物的制备原料包括化合物a和化合物c;
[0021]
所述化合物a的结构式为化合物c的结构式为
[0022]
所述r1、r3具有与上述相同的保护范围。
[0023]
优选地,所述化合物a和化合物c的摩尔比为(1~50):1,例如可以是1:1、5:1、10:
1、15:1、20:1、25:1、30:1、35:1、40:1、45:1或50:1等。
[0024]
作为本发明的优选技术方案,所述电致变色聚合物的制备原料还包括化合物b;
[0025]
所述化合物b的结构式为
[0026]
所述r2具有与上述相同的保护范围。
[0027]
优选地,所述化合物b和化合物c的摩尔比为(0.2~10):1,例如可以是0.2:1、0.5:1、1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1等。
[0028]
优选地,所述化合物a、化合物b和化合物c的摩尔比为(1~10):(0.2~5):1,例如可以是2:1:2、3:1:2、2:1:1、4:1:1、6:1:1、8:1:1、10:1:1、2:2:1、2:3:1、2:4:1、2:5:1、4:3:1、7:4:1或8:3:1等。
[0029]
作为本发明的优选技术方案,所述化合物c的制备方法采用如下方法制备得到,所述方法包括如下步骤:
[0030]
(1)化合物反应得到化合物
[0031]
(2)化合物与乙醛酸反应得到化合物
[0032]
(3)化合物与醇类化合物反应,得到所述化合物c。
[0033]
本发明中,通过设计化合物c的合成路线,对电致变色聚合物的结构进行设计,使电致变色聚合物具有较大的平面性,可以形成致密的分子堆积,且喹喔啉基团上具有的两个n原子具有较强的吸电子能力,通过在喹喔啉基团上引入单个烷氧基,可以有效的提升电致变色聚合物的溶解性,且制备得到的电致变色聚合物不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入。本发明提供的电致变色聚合物适用于作为电致变色材料,制备电致变色装置。
[0034]
本发明中,步骤(1)所述反应在保护气氛围和金属催化剂存在下进行,所述保护气包括氮气和氩气,所述金属催化剂包括锌粉;
[0035]
优选地,步骤(1)所述反应在溶剂存在下进行;
[0036]
所述溶剂为有机溶剂或有机溶剂与会的混合溶剂;所述有机溶剂选自乙酸、乙醇、丙醇、2-甲氧基乙醇中的任意一种或至少两种的组合。
[0037]
优选地,步骤(1)所述反应的温度为70~90℃,例如可以是70℃、72℃、74℃、76℃、
78℃、80℃、82℃、84℃、86℃、88℃或90℃等。
[0038]
优选地,步骤(1)所述反应后还包括后处理的步骤,所述后处理的方法包括抽滤、水洗、分液、洗涤。
[0039]
优选地,步骤(2)所述反应的温度为120~130℃,例如可以是120℃、121℃、122℃、123℃、124℃、125℃、126℃、127℃、128℃、129℃或130℃等。
[0040]
优选地,步骤(2)所述反应的时间为2~4h,例如可以是2h、2.5h、3h、3.5h或4h等。
[0041]
优选地,步骤(2)所述反应在溶剂存在下进行,所述溶剂选自乙醇、丙酮、异丙醇、n,n-二甲基甲酰胺或乙腈中的任意一种或至少两种的组合。
[0042]
优选地,步骤(3)中所述醇类化合物的碳原子个数为c1~c18,例如可以是c1、c2、c4、c6、c8、c10、c12、c14、c16或c18等。
[0043]
优选地,步骤(3)所述反应在催化剂存在下进行,所述催化剂选自三苯基膦、异辛醇或偶氮二甲酸二异丙酯中的任意一种或至少两种的组合。
[0044]
优选地,步骤(3)所述反应在溶剂存在下进行,所述溶剂选自四氢呋喃(thf)和/或乙醚;
[0045]
优选地,步骤(3)所述反应的温度为20~60℃,例如可以是20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃或60℃等。
[0046]
优选地,步骤(3)所述反应在惰性氛围中进行。
[0047]
优选地,所述化合物c的制备方法采用如下方法制备得到,所述方法包括如下步骤:
[0048]
(1)在惰性氛围、金属催化剂存在下,化合物在70~90℃下反应,抽滤、水洗、分液、洗涤,得到化合物
[0049]
(2)将化合物与乙醛酸,在120~130℃下,反应2~4h,得到化合物
[0050]
(3)在惰性氛围中,在催化剂存在下,将化合物与醇类化合物在室温下反应12~15反应,得到所述化合物c。
[0051]
第二方面,本发明提供一种如第一方面所述的电致变色聚合物的制备方法所述制
备方法包括如下步骤:
[0052]
将电致变色聚合物的制备原料混合后,进行聚合反应,得到所述电致变色聚合物。
[0053]
所述电致变色聚合物的制备原料包括化合物a和化合物c,所述反应的制备流程如下:
[0054][0055]
其中,r1、r3和n具有与上述相同的保护范围。
[0056]
所述电致变色聚合物的制备原料包括化合物a、化合物b和化合物c,所述反应的制备流程如下:
[0057][0058]
其中,r1、r2、r3、a、b、c、n具有与上述相同的保护范围。
[0059]
作为本发明的优选技术方案,所述聚合反应的温度为160~180℃,例如可以是160℃、162℃、164℃、166℃、168℃、170℃、172℃、174℃、176℃、178℃或180℃等。
[0060]
优选地,所述聚合反应的时间为4~48h,例如可以是4h、8h、12h、16h、20h、24h、28h、32h、36h、40h、44h或48h等。
[0061]
优选地,所述聚合反应在保护气氛围中进行,所述保护气包括氮气和氩气。
[0062]
优选地,所述聚合反应在有机溶剂存在下进行。
[0063]
需要说明的是,本发明对有机溶剂的选择没有任何特殊的限制,本领域常用的有机溶剂均适用,示例性地包括但不限于:n,n-二甲基乙酰胺(dmac)。
[0064]
优选地,所述聚合反应后还包括后处理的步骤,所述后处理的方法包括提纯。
[0065]
第三方面,本发明提供一种电致变色装置,所述电致变色装置包括依次设置的第一工作电极、电致变色层、电解质层、离子存储层和第二工作电极;
[0066]
所述电致变色层的材料由如第一方面所述的电致变色聚合物。
[0067]
优选地,所述电解质层的材料包括聚碳酸酯(pc)、聚(乙二醇)二丙烯酸酯(pegda)、0.2m锂双(三氟甲磺酰基)酰亚胺(litfsi)、二甲氧基2-苯基苯乙酮(bdk)。
[0068]
优选地,所述离子存储层是通过丝网印刷制备的聚(3,4-亚乙基二氧噻吩)
·
聚(苯乙烯磺酸盐)(pedot
·
pss)。
[0069]
优选地,所述第一工作电极和第二工作电极可以为ito玻璃。
[0070]
第四方面,本发明提供一种如第一方面所述的电致变色聚合物在制备智能窗、军
事伪装产品、调光调色产品、包装显示产品中的应用。
[0071]
优选地,所述制备智能窗、军事伪装产品、调光调色产品、包装显示产品的工艺包括涂布或印刷制程。
[0072]
与现有技术相比,本发明具有以下有益效果:
[0073]
(1)本发明中通过对电致变色聚合物结构的设计,进一步通过在喹喔啉基团上引入单个烷氧基,可以有效的提升电致变色聚合物的溶解性,且制备得到的电致变色聚合物不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入。本发明提供的电致变色聚合物适用于作为电致变色材料,制备电致变色装置。
[0074]
(2)由本发明提供的电致变色聚合物制备得到的电致变色装置具有优异的性能,其光学对比度较高为5.1%~18.1%,循环10万次后,对比度衰减率较低为5.9~17%,褪色时间较短为0.9~1.8s,着色时间较短为1.8~2.6s,着色效率较高为123.4~441.7cm2/c。
附图说明
[0075]
图1是本发明合成例1提供的化合物c1的核磁共振氢谱图;
[0076]
图2是本发明应用例1提供的电致变色装置的结构示意图;
[0077]
图3-5分别为应用例1-3提供的电致变色装置的循环伏安特性曲线;
[0078]
图6-8分别为对应用例1-3提供的电致变色装置的着色态和褪色态的光学调制率及其变色响应速度进行测试的测试结果图;
[0079]
图9-11分别为对应用例1-3提供的电致变色装置的稳定性进行测试的测试结果图;
[0080]
其中,1-第一工作电极,2-电致变色层,3-电解质层、4-离子存储层,5-第二工作电极。
具体实施方式
[0081]
下面结合附图并通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0082]
合成例1
[0083]
本合成例提供一种化合物c1及其制备方法,其制备方法如下:
[0084][0085]
(1)将化合物(100克,1当量)、锌粉(222.6克,10当量),乙酸(600毫升)、纯水(600毫升),加入到2升的三口瓶中,机械搅拌混合,氮气置换3次后,缓慢加热至80℃后,搅拌反应,进行点板,确定反应结束,停止加热,完成反应;
[0086]
然后将三口瓶移至常温水中冷却至室温后,将反应液缓慢倒入冰水中,并不停搅
拌;抽滤,得到白色固体,放置过夜;然后水洗3次,抽滤,将固体加入到1升的乙酸乙酯中溶解,抽滤得橙红色有机相,用饱和的碳酸氢钠溶液调节ph=8,进行分液,用纯水(1升)洗涤有机相2次,用饱和食盐水洗涤有机相1次,旋蒸、浓缩得到黄色固体72.4克(化合物),收率为47%;
[0087]
(2)在2l的四口烧瓶中,加入化合物(60克,0.206摩尔)、乙醛酸(质量百分含量50%的水溶液)(29.4克,0.206摩尔)、乙醇(1.5升),升温至125℃回流,反应2小时后,降温至室温,结束反应,然后进行抽滤,得到黄色固体,将黄色固体放入真空烘箱中在50℃下干燥5h,得到化合物51克,收率为76.89%;
[0088]
(3)在1l的三口烧瓶中加入化合物(29.6克,0.092摩尔)、三苯基膦(24克,0.092摩尔)、异辛醇(11.96克,0.092摩尔)、偶氮二甲酸二异丙酯(18.6克,0.092摩尔)、thf(700毫升),氮气置换三次,室温搅拌过夜,tlc点板检测反应。反应结束后,将反应液倒入冰水中,加入二氯甲烷萃取分液,有机相用饱和食盐水洗涤三次,加入无水硫酸钠干燥,旋干,得到粗品83克,纯石油醚作为淋洗剂,柱层析得到化合物c1 17.7克,收率为46.36%。
[0089]
对合成例1提供的化合物c1进行核磁共振氢谱测试:1h nmr(400mhz,chloroform-d)δ8.56(s,1h),7.85(s,1h),7.76(s,1h),4.55(s,2h),1.88(s,1h),1.39(s,9h),1.03
–
0.89(m,5h),其核磁共振氢谱图如图1所示。
[0090]
合成例2
[0091]
本合成例提供一种化合物c2及其制备方法,与合成例1的区别在于,将步骤(3)中异辛醇替换为等物质的量的正辛醇,其他条件与合成例1相同,得到化合物c2,收率为60.36%。
[0092]
合成例3
[0093]
本合成例提供一种化合物c3及其制备方法,与合成例1的区别在于,将步骤(3)中异辛醇替换为等物质的量的2-辛基十二醇,其他条件与合成例1相同,得到化合物c3,收率为35.78%。
[0094]
实施例1
[0095]
本实施例提供一种电致变色聚合物ptq1及其制备方法,其制备方法如下:
[0096][0097]
将化合物a(0.7克)、化合物c1(0.66克)、pd(oac)2(0.035克)、k2co3(0.65克)、pivoh(新戊酸,0.049克)、dmac(7毫升)加入到50毫升的schlenk管中,使用氮气置换3次,升温至170℃至出现回流,过夜反应24小时后,冷却至室温,加入少量的氯仿使固体充分溶解,将反应液缓慢倒入进150毫升的搅拌的甲醇溶液中,立即有固体析出,液体趋向于澄清呈黄色透明,加入3克乙二胺四乙酸(edta),体系变为黄色浊液,停止搅拌,抽滤,并用甲醇(100毫升)洗涤2次,得紫黑色固体(混edta),滤液为黄色浊液,分别用甲醇、丙酮、正己烷、氯仿索式提取后,旋蒸浓缩,缓慢滴入甲醇中沉淀,并不断搅拌,滤布过滤得所述紫黑色固体电致变色聚合物ptq1(1.02克),收率为58%。
[0098]
实施例2
[0099]
本实施例提供一种电致变色聚合物ptq2及其制备方法,其制备方法如下:
[0100][0101]
将化合物a(0.67克)、化合物b(0.30克)、化合物c1(0.43克)、pd(oac)2(0.035克)、k2co3(0.65克)、pivoh(0.049克)、dmac(7毫升)加入到50毫升的schlenk管中,氮气置换3次,梯度升温至170摄氏度至出现回流,过夜反应24小时后,冷却至室温,加入少量的氯仿使固体充分溶解,将反应液缓慢倒入进150毫升的不停搅拌的甲醇溶液中,立即有固体析出,液体趋向于澄清呈黄色透明,加入3克乙二胺四乙酸(edta),体系变为黄色浊液,停止搅拌,抽滤,并用甲醇(100毫升)洗涤2次,得紫黑色固体(混edta),滤液为黄色浊液,分别用甲醇、丙酮、正己烷、氯仿索式提取,将氯仿溶液旋蒸浓缩,缓慢滴入甲醇中沉淀,并不断搅拌,滤布过滤得紫黑色固体电致变色聚合物ptq2(0.81克),收率为58%。
[0102]
实施例3
[0103]
本实施例提供一种电致变色聚合物ptq3及其制备方法,其制备方法如下:
[0104][0105]
将化合物a(0.53克)、化合物b(0.36克)、化合物c1(0.25克)、pd(oac)2(0.035克)、k2co3(0.65克)、pivoh(0.049克)、dmac(7毫升)加入到50毫升的schlenk管中,氮气置换3次,梯度升温至170摄氏度至出现回流,过夜反应24小时后,冷却至室温,加入少量的氯仿使固体充分溶解,将反应液缓慢倒入进150毫升的不停搅拌的甲醇溶液中,立即有固体析出,液体趋向于澄清呈黄色透明,加入3克乙二胺四乙酸(edta),体系变为黄色浊液,停止搅拌,抽滤,并用甲醇(100毫升)洗涤2次,得紫黑色固体(混edta),滤液为黄色浊液,分别用甲醇、丙酮、正己烷、氯仿索式提取,将氯仿溶液旋蒸浓缩,缓慢滴入甲醇中沉淀,并不断搅拌,滤布过滤得紫黑色固体电致变色聚合物ptq3(0.56克),收率为49%。
[0106]
实施例4
[0107]
本实施例提供一种电致变色聚合物ptq4及其制备方法,与实施例2的区别仅在于,将化合物c1替换为等物质的量的化合物c2,其他条件与实施例2相同。
[0108]
实施例5
[0109]
本实施例提供一种电致变色聚合物ptq5及其制备方法,与实施例2的区别仅在于,将化合物c1替换为等物质的量的化合物c3,其他条件与实施例2相同。
[0110]
对比例1
[0111]
本对比例提供一种电致变色聚合物及其制备方法,与实施例2的区别仅在于,将化合物c1替换为等物质的量的化合物(购自苏州纳凯科技有限公司),其中r为乙基己基,其他条件与实施例2相同。
[0112]
对比例2
[0113]
本对比例提供一种电致变色聚合物及其制备方法,与实施例2的区别仅在于,将化合物c1替换为等物质的量的化合物(购自苏州纳凯科技有限公司),其中r为乙基己基,其他条件与实施例2相同。
[0114]
应用例1
[0115]
本应用例提供一种电致变色装置,所述电致变色装置包括依次设置的第一工作电极、电致变色层、电解质层、离子存储层和第二工作电极。
[0116]
所述第一工作电极和第二工作电极为ito玻璃;
[0117]
电致变色层的材料为电致变色聚合物ptq1;
[0118]
电解质层的材料为聚碳酸酯(pc)、聚(乙二醇)二丙烯酸酯(pegda)、0.2m锂双(三氟甲磺酰基)酰亚胺(litfsi)、二甲氧基2-苯基苯乙酮(bdk)的凝胶聚合物电解质;
[0119]
离子存储层的材料为聚(3,4-亚乙基二氧噻吩)
·
聚(苯乙烯磺酸盐)(pedot
·
pss)。
[0120]
所述电致变色装置的制备方法如下:
[0121]
(1)将两片ito玻璃清洗后通过o-plasma处理,得到第一工作电极和第二工作电极;
[0122]
(2)将电致变色聚合物ptq1溶解在二甲苯得到混合溶液(80毫克/毫升),然后对混合溶液进行机械过滤以除去杂质,通过静态旋涂(在600rpm的条件下旋涂6秒,然后在4000rpm下,旋涂30秒)至第一工作电极一侧,得到电致变色层,有效面积为2.5
×
3.0厘米;
[0123]
(3)将由聚碳酸酯(pc)、聚(乙二醇)二丙烯酸酯(pegda)、0.2m锂双(三氟甲磺酰基)酰亚胺(litfsi)、二甲氧基2-苯基苯乙酮(bdk)的凝胶聚合物电解质涂覆在电致变色层远离第一工作电极的一侧,得到电解质层;
[0124]
(4)将聚(3,4-亚乙基二氧噻吩)
·
聚(苯乙烯磺酸盐)通过丝网印刷至电解质层远离电致变色层的一侧,得到离子存储层;
[0125]
(5)将第二工作电极置于离子存储层远离电解质层的一侧,使用双面胶密封边缘,得到电致变色装置。
[0126]
应用例2-5
[0127]
应用例2-5分别提供一种电致变色装置,与应用例1的区别仅在于,将电致变色层的材料电致变色聚合物ptq1依次替换为电致变色聚合物ptq2-ptq5,其他条件与应用例1相同。
[0128]
使用辰华电化学工作站chi660e,对应用例1-3提供的电致变色装置的循环伏安特性曲线分别进行测试,其循环伏安特性曲线如图3-5所示。由图3-5可知,本发明提供的电致变色聚合物1-3都具有良好的电化学循环性能,具有一定的电荷储存能力。
[0129]
使用辰华电化学工作站chi660e和lambda 750分光光度计,对应用例1-3提供的电致变色装置的着色态和褪色态的光学调制率及其变色响应速度进行测试,其测试结果如图6-8所示。由图6-8可知,以本发明提供的电致变色聚合物制备得到的电致变色装置具有良好的光学调制率和较快的变色响应速度,其中,应用例3提供的电致变色装置,实现了0.9秒的褪色时间和1.8秒的着色时间。
[0130]
使用lambda 750分光光度计,对应用例1-3提供的电致变色装置的稳定性进行测试,其测试结果如图9-11所示。由图9-11可知,以本发明提供的电致变色聚合物制备得到的电致变色装置具有良好的循环稳定性。
[0131]
应用对比例1-2
[0132]
应用对比例1-2分别提供一种电致变色装置,与应用例1的区别仅在于,将电致变色层的材料电致变色聚合物1依次替换为应用对比例1-2提供的电致变色聚合物,其他条件与应用例1相同。
[0133]
对应用例1-5以及应用对比例1-2提供的电致变色装置的性能进行测试,测试方法如下:
[0134]
(1)起始电压:测试仪器:电化学工作站,测试方法:循环伏安法;测试条件:50mv/s,电压范围:2000mv~-2000mv,循环次数:8圈,电流设定范围:2.5
×
10-3
ma,起始电压取值:做曲线切线与y=0的交点。
[0135]
(2)最大吸收波长:测试仪器:lambda750联用,测试方法:吸收光谱,测试条件:光谱范围:400~800nm,取样间隔:1nm,最大吸收波长取值:分别测试上述装置着色态和透过态的透射率光谱,取透射率差值最大的波长为最大吸收波长。
[0136]
(3)光学对比度:测试仪器:电化学工作站与lambda750联用,测试仪器:电化学工作站与lambda750联用,测试方法:透过率时间谱,测试条件:电化学工作站:正向电压:褪色电压,时间:10s,反向电压:着色电压,时间:10s,lambda750:波长:最大吸收波长,时间:12分钟,取样间隔:0.2s,测试方式:透过(t%),取值:褪色态透过率-着色态透过率。
[0137]
(4)对比度衰减率:测试仪器:函数发生器+电化学工作站与lambda750联用,测试方法:在函数发生器上做阶跃循环,然后测试光学对比度,取值:(循环前的光学对比度-循环后的光学对比度)/循环前的光学对比度。
[0138]
(5)褪色时间:测试仪器:电化学工作站与lambda750联用,测试方法:透过率时间谱,测试条件:电化学工作站:正向电压:褪色电压,时间:10s,反向电压:着色电压,时间:10s,lambda750:波长:最大吸收波长,时间:12分钟,取样间隔:0.2s,测试方式:透过(t%),取值:由褪色态至着色态,透过率的变化达到光学对比度的95%时所需的时间。
[0139]
(6)着色时间:测试仪器:电化学工作站与lambda750联用,测试方法:透过率时间谱,测试条件:电化学工作站:正向电压:褪色电压,时间:10s,反向电压:着色电压,时间:10s,lambda750:波长:最大吸收波长,时间:12分钟,取样间隔:0.2s,测试方式:透过(t%),取值:由着色态至褪色态,透过率的变化达到光学对比度的95%时所需的时间。
[0140]
(7)着色效率:测试仪器:电化学工作站与lambda750联用,测试方法:透过率时间谱,测试条件:电化学工作站:正向电压:褪色电压,时间:10s,反向电压:着色电压,时间:10s,lambda750:波长:最大吸收波长,时间:12分钟,取样间隔:0.2s,测试方式:透过(t%),取值:q/a为x轴,以(log(t/tc))为y轴,作图,取切线的斜率为着色效率.q:单位时间内所通过的电流,a:装置的有效面积,t:透过率,tc:着色态的透过率,tb:褪色态透过率。
[0141]
应用例1-5以及对比应用例1s-2s提供的电致变色装置的性能测试结果如下表1所示:
[0142]
表1
[0143][0144]
由表1的内容可知,本发明中通过对电致变色聚合物结构的设计,进一步通过在喹喔啉基团上引入单个烷氧基,可以有效的提升电致变色聚合物的溶解性,且制备得到的电致变色聚合物不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入。由此制备得到的电致变色装置具有优异的性能,其光学对比度较高为5.1%~18.1%,循环10万次后,对比度衰减率较低为5.9~17%,褪色时间较短为0.9~1.8s,着色时间较短为1.8~2.6s,着色效率较高为123.4~441.7cm2/c。
[0145]
本发明中,进一步通过三种化合物(化合物a、化合物b和化合物c)作为电致变色聚合物的制备原料,可进一步提高电致变色聚合物的综合性能,由此电致变色聚合物制备得到的电致变色装置的起始电压为-0.25~0.17v,光学对比度较高为14.8%~18.1%,循环10万次后,对比度衰减率较低为5.9~9.6%,褪色时间较短为0.9~1.1s,着色时间较短为1.8~2.6s,着色效率较高为389.6~441.7cm2/c。
[0146]
与应用例2相比,若用于制备电致变色层的材料中电致变色聚合物中喹喔啉基团上具有两个烷氧基(对比应用例1)或者电致变色聚合物中喹喔啉基团上具有两个烷基(对比应用例2),制备得到的电致变色装置综合性能较差。
[0147]
综上所述,本发明中通过对电致变色聚合物结构的设计,进一步通过在喹喔啉基团上引入单个烷氧基,可以有效的提升电致变色聚合物的溶解性,且制备得到的电致变色聚合物不会产生较大的空间位阻,从而不会阻碍掺杂粒子的进入,由此制备得到的电致变色装置具有优异的性能。
[0148]
申请人声明,本发明通过上述实施例来说明本发明的详细结构特征和详细工艺流程,但本发明并不局限于上述详细结构特征和详细工艺流程,即不意味着本发明必须依赖上述详细结构特征和详细工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用部件的等效替换以及辅助部件的增加、具体方式的选择等,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1