一种基于微通道反应器合成吲哚-5-甲醛类化合物的方法与流程
1.本发明属于精细合成技术领域,涉及一种基于微通道反应器合成吲哚-5-甲醛类化合物的方法。
背景技术:
2.5-吲哚甲醛类化合物是重要的生化试剂和药物原料,由于5-吲哚甲醛含有活性较高的醛基,可方便的连接或转化为其它官能团,这使其成为合成众多吲哚类药物的重要原料,在临床医药的应用上,以5-吲哚甲醛为基础结构的衍生物被广泛应用于治疗各种疾病,并取得了卓越的疗效。例如治疗偏头痛的药物那拉曲坦(alexander w o,darko b,martin r o.indole derivative[p].gb2208646,1989.)。
[0003]
目前5-吲哚甲醛传统工艺合成路线为以较便宜的3-甲基-4-硝基-苯甲酸为原料,经酯化(hidenori n,xingguo l,hiroyuki a.effect of the ortho modification of azobenzene on the photoregulatory efficiency of dna hybridization and the thermal stability of its cis form[j].chem.eur.j..2010,16:2054-2062.)、胺化(showalter h d,li s.concise syntheses of the novel 1h-pyrrolo[3,2-g]quinazoline ring system and its[2,3-f]angular isomer[j].j.org.chem..1996,61:1155-1158.)、脱水(krishnappa m,mohamed ap.rapid method of converting primary amides to nitriles and nitriles to primary amides by zncl2 using microwaves under different reaction conditions[j].synthetic communications..2007,37:1545-1550.),再用l-b法合成吲哚环(batcho a d,leimgruber w.intermediates for indoles[p],us3976639,1976.),最后还原氰基得到5-吲哚甲醛(hojatollah m,abbas s.synthesis of carbon-14analogue of 1,5diaryl-5-[14c]-1,2,3-triazoles[j].applied radiation and isotopes..2004,60:665-668.)。这一方法原料成本低,提纯简单,但反应步骤较多,排放三废也多,导致总成本过高。另外还有5-溴吲哚直接用锂试剂锂化,然后与dmf反应(mikelp m,johnf s,henry r.metal-halogen exchange of bromoindoles.a route to substituted indoles[j].j.org.chem..1986,51(26):5106-10.),该方法的优点是合成路线较短,一步就能合成最终产物。但缺点也很明显,首先所使用的kh,t-buli较为活泼,工业化批量生产比较危险。其次,反应在-78℃的低温条件下进行,这一温度在工业上虽然可以用液氮浴实现,但成本较高,且比较危险。还有pd催化的氰基化反应,还原氰基生成醛基(fujikawa y,urano y,komatsu t.design and synthesis of highly sensitive fluorogenic substrates for glutathione s-transferase and application for activity imaging in living cells[j].journal of the american chemical society..2008,130(44):14533-14543.),反应以5-溴吲哚为原料,经过氰基化,形成5-氰基吲哚,然后再将氰基还原为醛基,反应需要使用剧毒品。
技术实现要素:
[0004]
本发明的目的就是为了提供一种基于微通道反应器合成吲哚-5-甲醛类化合物的方法。
[0005]
本发明的目的可以通过以下技术方案来实现:
[0006]
一种基于微通道反应器合成吲哚-5-甲醛类化合物的方法,其特征在于,包括以下步骤:
[0007]
(1)往充满氮气的烧瓶中加入卤代吲哚类化合物和四氢呋喃,充分溶解,再加入混合后的烷基锂和攫锂剂搅拌均匀;
[0008]
(2)往另一充满氮气的烧瓶中加入甲酸酯的四氢呋喃溶液;
[0009]
(3)将步骤(1)和步骤(2)所得物料泵入充满氮气的微通道反应器中进行甲酰化反应,所得完成反应的物料淬灭、萃取、浓缩结晶,即得到目标产物吲哚-5-甲醛类化合物;
[0010]
所述卤代吲哚类化合物的化学结构式为:
[0011][0012]
其中,r为h、c1-c10的烷基、卤素、烷氧基或氨基;
[0013]
x选自cl、br、i、磺酸酯,且当x为磺酸酯时,r为卤素。
[0014]
进一步的,本发明的合成工艺路线如下:
[0015][0016]
进一步的,r为甲基、乙基、丙基、异丙基、叔丁基、卤素、氨基、烷氧基。
[0017]
进一步的,x可选为br。
[0018]
进一步的,所述烷基锂为甲基锂、丁基锂、仲丁基锂、叔丁基锂中一种或者几种的组合。
[0019]
进一步的,所述攫锂剂选自含氮、含氧的、可以与li离子配位的多齿有机化合物,其选自乙二醇二甲醚、四甲基乙二胺、12-冠-4醚中的至少一种,可选为12-冠-4醚。
[0020]
进一步的,所述甲酸酯选自甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、甲酸苯酯中一种,可选为甲酸丁酯。
[0021]
进一步的,甲酰化反应过程中的温度为-60℃~90℃,可选为-20℃~50℃,反应物料在微通道反应器中的停留时间为40~120s,可选为90s。
[0022]
进一步的,卤代吲哚类化合物、烷基锂、攫锂剂、甲酸酯物质的量的配比为1:(1.5~3.0):(1.5~3.0):(2.0~4.0),可选为1:2.0:2.0:1.5。
[0023]
进一步的,微通道反应器的总流速大于等于30ml/min,且步骤(1)和步骤(2)所得物料的流速均维持为15ml/min~40ml/min。
[0024]
进一步的,步骤(1)所得物料中,卤代吲哚类化合物的浓度为0.5~2.0mol/l,可选为1.0mol/l,烷基锂和攫锂剂的浓度分别为1.0~2.0mol/l,可选为1.5mol/l;
[0025]
步骤(2)所得物料中,甲酸酯浓度为1.0~2.0mol/l,可选为1.5mol/l。
[0026]
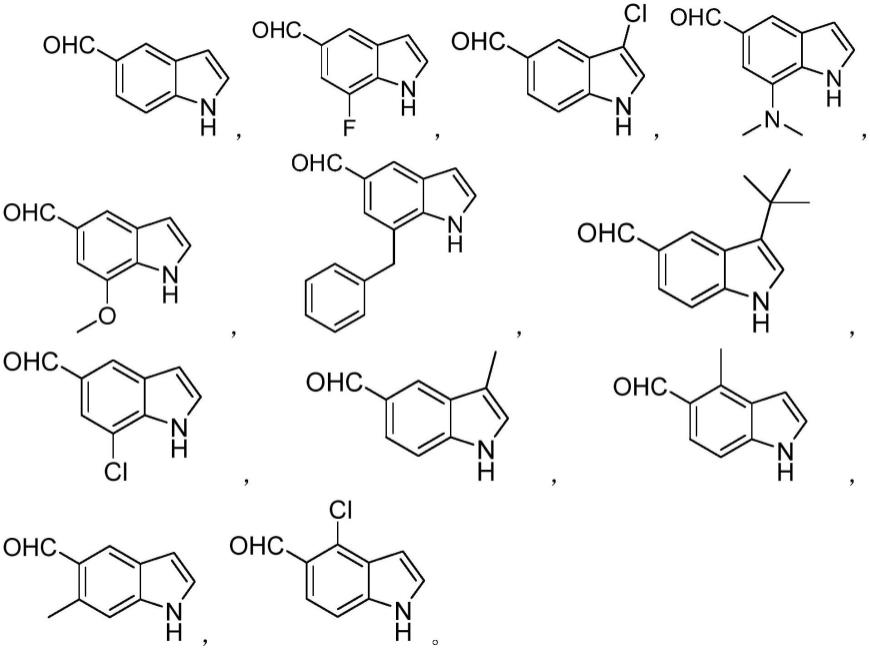
进一步的,所得吲哚-5-甲醛类化合物选自以下化合物的任一种:
[0027][0028][0029]
本发明创新之一是使用攫锂剂对烷基锂进行处理,利用攫锂剂强大的配位能力,与金属锂离子络合,打破或者削弱锂离子与烷基的静电结合,释放更多的烷基负离子,夺取吲哚环上卤素或者磺酸酯基形成吲哚阴离子中间体,进一步与甲酸酯进行亲核加成-消除反应得到吲哚甲醛,攫锂剂的加入有效提高了反应活性,加速完成卤代吲哚环的亲核取代反应,提高反应活性。本发明创新之二是使用微通道反应器进行吲哚甲醛的合成,物料接触时间短、混合均匀、有利于反应进行,容易控制反应热交换,反应可以连续化进行。
[0030]
与现有技术相比,本发明具有以下优点:
[0031]
(i)本发明合成吲哚-5-甲醛类化合物的合成路线简捷,采用连续化一锅煮合成工艺,可以连续生产。
[0032]
(ii)采用微通道反应器,能够实现对反应过程的精准控制,最终能够制得高纯度的化合物,并且大幅度地提高了收率。
[0033]
(ii)本发明的连续合成吲哚-5-甲醛类化合物的工艺,单个微单元反应的平均时间仅为几十秒,相较于釜式间歇式工艺的几十分钟,大幅度提高了反应效率。
[0034]
(v)本发明的连续合成的微通道反应系统,将原本大剂量的反应切割为多个微单元反应,实现了小剂量反应,整个反应过程为连续化,避免了由于局部导热不畅通导致飞温危险,反应可以在-60℃~90℃区间内进行,从而提高了反应的安全性。
[0035]
微通道反应器技术已逐渐成为国际精细化工技术领域的研究热点。微通道反应器是一种借助于特殊微加工技术以固定基质制造的可用于化学反应的三维结构元件。微通道反应器通常含有很小的通道尺寸(当量直径小于500μm)和通道多样性,流体在这些通道中
流动、混合、反应。因此在这种微构造的化学设备中具有极大的比表面积(表面积/体积)。由此带来的优势是极大的传质和传热效率,即能实现对反应温度的精确控制和对反应物料以精确配比瞬间混合,提高收率选择性、安全性、以及产品质量,同时该设备体积小、能耗低。
具体实施方式
[0036]
为了使本发明的目的、技术方案及优点更加清楚明白,下面结合具体实施例等对本发明进行详细说明。本实施例以本发明技术方案为前提进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0037]
下面先对本发明所采用的合成工艺进行说明:
[0038]
本发明提供的一种基于微通道反应器合成吲哚-5-甲醛类化合物的方法,其合成工艺路线如下:
[0039][0040]
具体可包括以下步骤:
[0041]
(1)往充满氮气的烧瓶中加入卤代吲哚类化合物和四氢呋喃,充分溶解,再加入混合后的烷基锂和攫锂剂搅拌均匀;
[0042]
(2)往另一充满氮气的烧瓶中加入甲酸酯的四氢呋喃溶液;
[0043]
(3)将步骤(1)和步骤(2)所得物料泵入充满氮气的微通道反应器中,进行甲酰化反应,所得完成反应的物料淬灭、萃取、浓缩结晶,即得到目标产物吲哚-5-甲醛类化合物;
[0044]
所述卤代吲哚类化合物的化学结构式为:
[0045][0046]
其中,r为h、c1-c10的烷基、卤素、烷氧基或氨基;
[0047]
x选自cl、br、i、磺酸酯,且当x为磺酸酯时,r为卤素。
[0048]
在一些具体的实施例中,r为甲基、乙基、丙基、异丙基、叔丁基、卤素、氨基、烷氧基。
[0049]
在一些具体的实施例中,x可选为br。
[0050]
在一些具体的实施例中,所述烷基锂为甲基锂、丁基锂、仲丁基锂、叔丁基锂中一种或者几种的组合。
[0051]
在一些具体的实施例中,所述攫锂剂选自含氮、含氧的、可以与li离子配位的多齿有机化合物,其选自乙二醇二甲醚、四甲基乙二胺、12-冠-4醚中的至少一种,可选为12-冠-4醚。
[0052]
在一些具体的实施例中,所述甲酸酯选自甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸丁酯、甲酸苯酯中一种,可选为甲酸乙酯。
[0053]
在一些具体的实施例中,甲酰化反应过程中的温度为-60~90℃,可选为-20℃~
溴吲哚0.1mol和四氢呋喃100ml,充分溶解;正丁基锂0.3mol(2.0m,150ml)和12-冠-4醚0.3mol混合后搅拌后加入到5-溴吲哚底物中,接入流速为(20ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸乙酯0.3mol,四氢呋喃200ml配成溶液,接入流速(20ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为0℃,使物料流入微通道反应器,物料在微通道反应器内停留时间为72s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:3:3:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将物料加入到100g冰水中淬灭,旋蒸回收四氢呋喃后再加入石油醚萃取,浓缩后结晶析出产物,收率为90%。
[0065]1h nmr(dmso-d6):11.66(br,1h),10.00(s,1h),8.22(s,1h),7.67(d,1h,,j=7.9),7.60(br,1h),7.57(d,1h,j=7.6),6.70(br,1h)。
[0066]
实施例2
[0067]
操作同实施例1。仅仅调整反应物质比例为底物:烷基锂:攫锂剂:甲酸酯比例为1:2:2:2,反应收率为82%。
[0068]
实施例3c1常规合成
[0069]
反应物种类和比例同实施例1。向配有机械搅拌、惰性气体保护、恒温油浴、恒压加料漏斗、取样口的1.0l的圆底烧瓶中,吹入干燥的n2,并在n2保护下加入5-溴吲哚的thf溶液,搅拌,滴入丁基锂与12-冠-4醚的thf溶液,维持瓶内温度为0℃,搅拌反应30min后缓缓滴入甲酸酯的thf溶液,每隔0.5h从取样口取样进行tlc分析,待5-溴吲哚点消失认为反应完成,反应5.0h反应完成。缓缓加入100g冰水淬灭反应,搅拌15min后旋蒸回收thf,剩余物质中的液体用石油醚萃取,剩余物质中固体物质与萃取有机相合并,重结晶得到产品,收率80%。
[0070]
实施例4
[0071]
化合物c2的合成
[0072]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入7-氟-5-碘吲哚0.1mol和四氢呋喃100ml,充分溶解;甲基锂0.25mol(2.0m,125ml)和乙二醇二甲醚(dme)0.2mol,混合搅拌后加入到底物溶液中,接入流速为(20ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸丁酯(0.3mol,溶解于200ml四氢呋喃)溶液,接入流速(20ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为40℃,使物料泵入微通道,停留时间为72s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:2.5:2:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,旋蒸回收四氢呋喃后再加入石油醚萃取,浓缩后结晶析出产物,收率为78%。
[0073]1h nmr(cdcl3):10.0(s,1h),8.76(br,1h),7.97(s,1h),7.47(dd,j=11.0,1h),7.35(m,1h),6.76(m,1h).
[0074]
实施例5
[0075]
操作同实施例4。仅仅调整反应温度为60℃,反应收率为67%。
[0076]
实施例6c2的常规合成
[0077]
合成装置与操作同实施例3。反应底物以及比例同实施例4,反应耗时5.0h,收率61%。
[0078]
实施例7
[0079]
化合物c3的合成
[0080]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入3-氯-5-磺酸酯基吲哚0.1mol和四氢呋喃80ml,充分溶解;叔丁基锂0.15mol(1.0m,150ml)和四甲基乙二胺(tmeda)0.15mol混合后搅拌后加入到底物中,接入流速为(25ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸甲酯(0.3mol溶解于200ml四氢呋喃)溶液,接入流速(25ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为-15℃,使物料泵入微通道反应器,停留时间为58s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:1.5:1.5:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,旋蒸回收thf,再加入石油醚萃取,浓缩后结晶析出产物,收率为90%。
[0081]1h nmr(cdcl3):δ=10.08(s,1h),8.66(bs,1h),8.18(s,1h),7.83(dd,j1=8.6hz,j2=1.4hz,1h),7.47(d,j=8.6hz,1h),7.31(d,j=2.5hz,1h).
[0082]
实施例8
[0083]
同实施例7。仅仅调整反应温度为0℃,反应收率为81%。
[0084]
实施例9
[0085]
同实施例7。仅仅调整反应温度为室温,反应收率为71%。
[0086]
实施例10
[0087]
同实施例7。仅仅调整反应温度为40℃,反应收率为60%。
[0088]
实施例11
[0089]
化合物c4的合成
[0090]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入7-二甲氨基-5-溴吲哚0.1mol和四氢呋喃150ml,充分溶解;正丁基锂0.25mol(2.0m,125ml)和tmeda 0.1mol混合后搅拌后加入到底物中,接入流速为(15ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸丙酯(0.4mol,250ml四氢呋喃)溶液,接入流速(15ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为室温,使物料流入微通道,停留时间为100s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:2.5:1:4,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,旋蒸回收thf,再加入石油醚萃取,浓缩后结晶析出产物,收率为77%。
[0091]1h nmr(400mhz,cdcl3):δ9.9(s,1h),9.88(s,1h),8.19(s,1h),7.14(d,j=8.2hz,1h),6.96(s,1h),6.56(d,j=8.2hz,1h),3.01(s,6h).
[0092]
实施例12
[0093]
化合物c5的合成
[0094]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入7-甲氧基-5-氯吲哚0.1mol和四氢呋喃200ml,充分溶解;叔丁基锂0.25mol(1.0m,250ml)和dme 0.15mol混合后搅拌后加入到底物中,接入流速为(30ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸丁酯(0.2mol和200ml四氢呋喃)溶液,接入流速(15ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为-20℃,使物料泵入微通道,停留时间为64s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:2.5:1.5:2.0,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,旋蒸回收thf,再加入石油醚萃取,浓缩后结晶析出产物,收率为87%。
[0095]1h nmr(400mhz,cdcl3):δ=10.2(s,1h),9.87(s,1h),8.56(s,1h),7.29(s,1h),
7.16(d,j=7.5hz,1h),6.55(d,j=7.4hz,1h),3.97(s,3h)。
[0096]
实施例13
[0097]
操作同实施例12。仅仅调整反应温度为5℃,反应收率为80%。
[0098]
实施例14
[0099]
操作同实施例13。仅仅调整攫锂剂为12-冠-4,反应收率为88%。
[0100]
实施例15常规合成c5
[0101]
装置与反应工艺流程同实施例3。物料投料以及配比、反应温度同实施例12,tlc检测底物消耗完毕耗时7.5h,产品收率62%。
[0102]
实施例16
[0103]
化合物c6的合成
[0104]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入7-苄基-5-碘吲哚0.1mol和四氢呋喃75ml,充分溶解;叔丁基锂0.3mol(1.0m,300ml)和12冠-4-醚0.3mol混合后搅拌后加入到底物中,接入流速为(15ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸苯酯(0.3mol和四氢呋喃350ml)溶液,接入流速(15ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为室温,使物料泵入微通道,停留时间为100s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:3:3:3,持续5分钟使物料充满微通道,5分钟后开始接收物料,将完成反应的物料加入到冰水中淬灭,旋蒸回收thf,再加入石油醚萃取,浓缩后结晶析出产物,收率为78%。
[0105]1h nmr(400mhz,cdcl3):δ=10.3(s,1h),9.87(s,1h),8.74(s,1h),7.58(s,1h),7.26(m,6h),6.51(d,j=8.2hz,1h),4.01(s,2h).
[0106]
实施例17
[0107]
操作同实施例16。仅仅改变反应温度为-15℃,反应收率为85%。
[0108]
实施例18常规合成c6
[0109]
装置与反应工艺流程同实施例3。物料投料以及配比、反应温度同实施例16,tlc检测底物消耗完毕耗时6.0h,产品收率60%。
[0110]
实施例19
[0111]
化合物c7的合成与表征
[0112]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入3-叔丁基-5-溴吲哚0.1mol和150ml四氢呋喃,充分溶解;甲基锂0.15mol(2.0m,75ml)和0.2mol的dme混合后搅拌后加入到底物中,泵入流速为(20ml/min)的进样泵,向另外充满氮气的烧瓶中先加入0.2mol的甲酸丁酯和220ml四氢呋喃的溶液,泵入流速(20ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为室温,使物料流入微通道反应器,停留时间为75s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:1.5:2:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,再加入石油醚萃取,浓缩后结晶析出产物,收率为80%。
[0113]1h nmr(400mhz,cdcl3):δ=10.8(s,1h),9.82(s,1h),7.76(d,j=7.4hz,2h),7.74(d,j=7.5hz,1h),7.20(s,1h),1.37(s,9h)。
[0114]
实施例20
[0115]
操作同实施例19,仅仅改变攫锂剂为tmeda,反应收率为88%。
[0116]
实施例21常规合成c7
[0117]
装置与反应工艺流程同实施例3。物料投料以及配比、反应温度同实施例19,tlc检测底物消耗完毕耗时5.0h,产品收率71%。
[0118]
实施例22
[0119]
化合物c8的合成
[0120]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入7-氯-5-磺酸酯基吲哚0.1mol和四氢呋喃80ml,充分溶解;叔丁基锂0.15mol(1.0m,150ml)和tmeda 0.2mol混合后搅拌后加入到底物中,接入流速为(15ml/min)的进样泵,向另外充满氮气的烧瓶中先加入甲酸乙酯0.2mol和250ml四氢呋喃的溶液,接入流速(15ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为-10℃,使物料泵入微通道反应器,停留时间为100s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:1.5:2:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,再加入石油醚萃取,浓缩后结晶析出产物,收率为72%。
[0121]1h nmr(cdcl3):δ=10.00(s,1h),8.60(br,1h),8.08(s,1h),7.79(d,j)1.3hz,1h),7.37(dd,j)3.2,2.4hz,1h),6.77(dd,j)3.3,2.3hz,1h).
[0122]
实施例23
[0123]
操作同实施例22,仅仅改变反应温度为0℃,反应收率为82%。
[0124]
实施例24c8常规合成
[0125]
装置与反应工艺流程同实施例3.物料投料以及配比、反应温度同实施例22,tlc检测底物消耗完毕耗时6.0h,产品收率63%。
[0126]
实施例25
[0127]
化合物c12的合成
[0128]
首先向微通道中通入氮气,使体系内充满氮气,接着向充入氮气的烧瓶中加入4-氯-5-磺酸酯基吲哚0.1mol和四氢呋喃150ml,充分溶解;正丁基锂0.15mol(2.0m,75ml)和dme 0.2mol混合后搅拌后加入到底物中,泵入流速为(20ml/min)的进样泵,向另外充满氮气的烧瓶中先加入0.3mol的甲酸乙酯和220ml的四氢呋喃溶液,接入流速(20ml/min)的进样泵,准备工作完成后,同时开始两个泵的开关,反应温度为10℃,使物料流入微通道,停留时间为72s,其中底物:烷基锂:攫锂剂:甲酸酯比例为1:1.5:2:3,持续5分钟使物料充满微通道,7分钟后开始接收物料,将完成反应的物料加入到100g冰水中淬灭,再加入石油醚萃取,浓缩后结晶析出产物,收率为75%。
[0129]1h nmr(dmso-d6)δ10..50(s,1h),10.2(s,1h),8.03(m,1h),7.65(d,j=8.6hz,1h),7.20(d,j=7.7hz,1h),6.83(d,j=8.0hz,1h).
[0130]
实施例26
[0131]
操作同实施例25,仅仅改变攫锂剂为tmeda,反应收率为84%。
[0132]
实施例27c12常规合成
[0133]
装置与反应工艺流程同实施例3。物料投料以及配比、反应温度同实施例22,tlc检测底物消耗完毕耗时5.5h,产品收率61%。
[0134]
实施例28
[0135]
操作同实施例1,变化是仅仅不加攫锂剂。每隔2min进行tlc检测,发现未有产物生
成,所得物料再次泵入微通道反应器,流出物中依然没有目标产物。
[0136]
实施例29
[0137]
操作同实施例3,变化是仅仅不加攫锂剂。加入甲酸酯后每隔15min进行tlc检测,发现未有产物生成,搅拌5.0h,未发现有目标产物生成。
[0138]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1