一种活性氧响应型两亲性聚合物及其制备方法和作为活性氧靶向的疏水药物递送载体的应用与流程
1.本发明涉及药物载体技术领域,特别涉及一种活性氧响应型两亲性聚合物及其制备方法和作为活性氧靶向的疏水药物递送载体的应用。
背景技术:
2.两亲性聚合物形成的纳米胶束是一种新型的材料,在生物医药、农业、工业涂料等多个领域具有巨大的潜在应用价值。纳米胶束的尺寸较小,因而具有非常好的细胞膜穿透能力,因此在生物医药领域的研究受到人们的广泛关注。两亲性聚合物形成的纳米胶束具有疏水性内核,可以包裹疏水性药物,将其递送至病变部位,可以大大提高药物,特别是非水溶性药物的利用率。
3.传统方法制备的纳米胶束主要是通过将疏水单体和亲水单体共聚形成两亲性聚合物,通过改变疏水/亲水单体之间的比例来调控纳米胶束的尺寸、临界浓度等物理性质。这种方法制备的纳米胶束其对药物的释放并不可控,其有可能在无需治疗的位置释放药物,在非病变位置导致药物的副作用,同时病变位置药物的有效浓度降低。
技术实现要素:
4.有鉴于此,本发明目的在于提供一种活性氧响应型两亲性聚合物及其制备方法和作为活性氧靶向的疏水药物递送载体的应用,本发明提供的活性氧响应型两亲性聚合物具有活性氧响应特性,能够在含有活性氧的病变部位靶向释放药物。
5.为了实现上述发明目的,本发明提供以下技术方案:
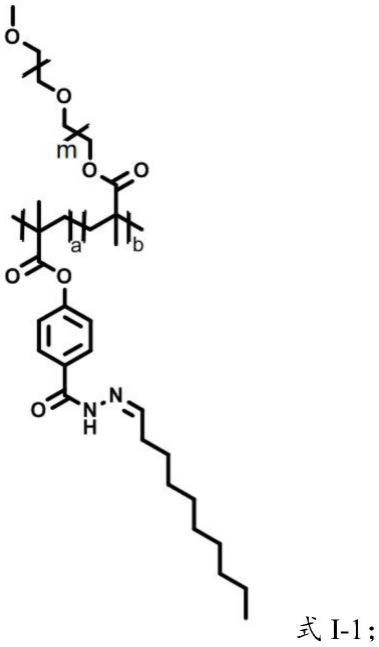
6.本发明一种活性氧响应型两亲性聚合物,具有式i所示结构:
[0007][0008]
式i中,a为苯环或-(ch2)
k-;
[0009]
a=1~160,b=1~160,m=10~90,n=4~20,k=1~10。优选的,具有式i-1所示结构:
[0010]
[0011]
式i-1中,a=1~160,b=1~160,m=10~90。
[0012]
本发明提供了上述活性氧响应型两亲性聚合物的制备方法,包括以下步骤:
[0013]
具有式c所示结构含羟基的酰肼与具有式d所示结构的疏水醛进行wolff
–
kishner反应,得到具有式e所示结构的腙结构中间体;
[0014][0015]
具有式e所示结构的腙结构中间体与甲基丙烯酰氯进行取代反应,得到具有式f所示结构的腙结构单体;
[0016][0017]
在引发剂的作用下,具有式f所示结构的腙结构单体与具有式g所示结构的聚乙二醇单甲醚甲基丙烯酸酯进行自由基聚合反应,得到具有式i所示结构的活性氧响应型两亲性聚合物;
[0018][0019]
优选的,所述具有式c所示结构含羟基的酰肼为4-羟基苯甲酰肼、羟基已酰肼或3-羟基丙酰肼;
[0020]
所述具有式d所示结构的疏水醛为正癸醛。
[0021]
优选的,所述wolff
–
kishner反应的温度为60~80℃,时间为2~12h;
[0022]
所述取代反应的温度为室温,时间为2~16h;
[0023]
所述自由基聚合反应的温度为60~80℃,时间为24~72h。
[0024]
本发明提供了上述活性氧响应型两亲性聚合物作为活性氧靶向的疏水药物递送载体的应用。
[0025]
本发明提供了上述一种载药纳米胶束,包括上述活性氧响应型两亲性聚合物,和被所述活性氧响应型两亲性聚合物分子链段包裹的疏水药物。
[0026]
优选的,所述疏水药物为姜黄素、阿霉素、富马酸二甲酯和酮洛芬中的一种或几种;
[0027]
所述活性氧响应型两亲性聚合物与疏水药物的质量比为10000:1~10。
[0028]
优选的,所述纳米胶束的粒径为6~20nm。
[0029]
本发明提供了上述载药纳米胶束的制备方法,包括以下步骤:
[0030]
将活性氧响应型两亲性聚合物、疏水药物与醇溶剂混合,去除醇溶剂,得到混合组分;
[0031]
将所述混合组分与水相液体超声混合,得到载药纳米胶束。
[0032]
本发明提供了一种活性氧响应型两亲性聚合物,具有式i所示结构。本发明提供的活性氧响应型两亲性聚合物含有腙结构,本发明通过腙结构链接聚合物骨架与疏水部分,当聚合物所形成的胶束接触病变部位(如伤口、炎症部位)的活性氧时,腙结构发生断裂,使得聚合物的亲水部分与疏水部分分离,亲水部分会溶解于水中,使得胶束解体。随着胶束的解体,其内包裹的药物可以释放至活性氧水平较高的位置,实现定点治疗。本发明以活性氧响应型两亲性聚合物作为疏水药物载体,对细胞中的活性氧有较好的响应,在保持聚合物纳米胶束本身优势的基础上,进一步增加了其内部包裹的药物的响应型释放,降低了药物在非病变部位提前释放的风险,同时提高了病变部位的有效浓度,而在病变部位的活性氧被清除后又可以停止继续释放,避免了药物的毒副作用。同时,本发明提供的活性氧响应型两亲性聚合物具有两亲性结构,其作为药物载体能够克服在疏水药物水中溶解度低的缺陷,极大扩展了疏水药物的适用范围。
[0033]
本发明提供了上述活性氧响应型两亲性聚合物的制备方法,此法操作简单,成本低廉,反应条件温和,适合工业化批量生产。
附图说明
[0034]
图1为实施例1中活性氧响应型两亲性聚合物的合成路线;
[0035]
图2为4-羟基癸亚胺苯甲酰肼的1h-nmr谱图;
[0036]
图3为聚合物单体的1h-nmr谱图;
[0037]
图4为活性氧响应型两亲性聚合物的1h-nmr谱图;
[0038]
图5为纳米胶束粒径的粒径分布;
[0039]
图6为纳米胶束粒径稳定性测试结果;
[0040]
图7为纳米胶束荧光强度测试结果;
[0041]
图8为fox标准溶液的标准曲线;
[0042]
图9为纳米胶束的抗氧化测试结果;
[0043]
图10为不同浓度下细胞的存活率;
[0044]
图11为药物释放测试结果;
[0045]
图12为细胞内ros清除测试结果。
具体实施方式
[0046]
本发明提供了一种活性氧响应型两亲性聚合物,具有式i所示结构:
[0047][0048]
在本发明中,式i中,a为苯环或-(ch2)
k-。
[0049]
在本发明中,a、b表示重复单元的数量,其中,a=1~160,优选为5~150,更优选为10~120,进一步优选为50~100;b=1~160,优选为5~150,更优选为10~120,进一步优选为50~100;m=10~90,优选为20~80,更优选为30~50;n=8;k=1~10,优选为1~5,更优选为2~4。
[0050]
在本发明中,所述活性氧响应型两亲性聚合物优选具有式i-1所示结构:
[0051][0052]
式i-1中,a=1~160,优选为5~150,更优选为10~120,进一步优选为50~100;b=1~160,优选为5~150,更优选为10~120,进一步优选为50~100;m=10~90,优选为20~80,更优选为30~50。
[0053]
在本发明中,所述活性氧响应型两亲性聚合物的数均分子量优选为10000~200000,更优选为50000~100000。
[0054]
本发明提供了上述活性氧响应型两亲性聚合物的制备方法,包括以下步骤:
[0055]
具有式c所示结构含羟基的酰肼与具有式d所示结构的疏水醛进行wolff
–
kishner反应,得到具有式e所示结构的腙结构中间体;
[0056][0057]
具有式e所示结构的腙结构中间体与甲基丙烯酰氯进行取代反应,得到具有式f所示结构的腙结构单体;
[0058][0059]
在引发剂的作用下,具有式f所示结构的腙结构单体与具有式g所示结构的聚乙二醇单甲醚甲基丙烯酸酯进行自由基聚合反应,得到具有式i所示结构的活性氧响应型两亲性聚合物;
[0060][0061]
在本发明中,具有式c所示结构含羟基的酰肼与具有式d所示结构的疏水醛进行wolff
–
kishner反应,得到具有式e所示结构的腙结构中间体。在本发明中,所述具有式c所示结构含羟基的酰肼与具有式d所示结构的疏水醛的摩尔比优选为1~3:1~3,更优选为1:1。在本发明中,所述具有式c所示结构含羟基的酰肼优选为4-羟基苯甲酰肼、羟基已酰肼或3-羟基丙酰肼;所述具有式d所示结构的疏水醛优选为正癸醛。
[0062]
在本发明中,所述wolff
–
kishner反应的反应溶剂优选为醇溶剂,所述醇溶剂优选为甲醇和/或乙醇。在本发明中,所述wolff
–
kishner反应的温度优选为60~80℃,更优选为65~78℃;时间优选为2~12h,更优选为4h。在本发明中,所述wolff
–
kishner反应后,本发明优选去除所得wolff
–
kishner反应液的醇溶剂;所述去除醇溶剂的方式优选为旋蒸。
[0063]
在本发明中,具有式e所示结构的腙结构中间体与甲基丙烯酰氯进行取代反应,得到具有式f所示结构的腙结构单体。在本发明中,所述具有式e所示结构的腙结构中间体与甲基丙烯酰氯的摩尔比优选为1:1~3,更优选为1:1.1。
[0064]
在本发明中,所述取代反应优选在缚酸剂的存在下进行,所述缚酸剂优选为三乙胺。在本发明中,所述具有式f所示结构的腙结构中间体与缚酸剂的摩尔比优选为1:1~4,更优选为1:1.3。
[0065]
在本发明中,所述取代反应使用的有机溶剂优选为乙酸乙酯、二氯甲烷和四氢呋喃中的一种或几种。在本发明中,所述取代反应的温度优选为室温,时间优选为2~16h,更优选为5~10h。
[0066]
所述取代反应后,本发明优选对所得取代反应液进行后处理,所述后处理优选包括:
[0067]
对所述取代反应液进行固液分离,所得液体去除有机溶剂后进行柱层析分离,得
到具有式f所示结构的腙结构单体纯品。
[0068]
在本发明中,所述固液分离的方式优选为过滤;所述去除有机溶剂的方式优选为旋蒸。在本发明中,所述柱层析分离的流动相优选为乙酸乙酯和己烷,所述乙酸乙酯和己烷的体积比优选为1:1。
[0069]
在本发明中,在引发剂的作用下,具有式f所示结构的腙结构单体与具有式g所示结构的聚乙二醇单甲醚甲基丙烯酸酯进行自由基聚合反应,得到具有式i所示结构的活性氧响应型两亲性聚合物。在本发明中,所述引发剂优选为偶氮类引发剂,进一步优选为偶氮二异丁腈。在本发明中,所述具有式f所示结构的腙结构单体与具有式g所示结构的聚乙二醇单甲醚甲基丙烯酸酯的摩尔比优选为1:0.5~3,更优选为1:1~2。
[0070]
在本发明中,所述自由基聚合反应优选在有机溶剂中进行,所述有机溶剂优选为四氢呋喃。在本发明中,所述自由基聚合反应的温度优选为60~80℃,更优选为65~75℃;时间优选为24~72h,更优选为48~60h。
[0071]
在本发明中,所述自由基聚合反应后,本发明优选对所得自由基聚合反应液进行后处理,所述后处理优选包括:
[0072]
去除所述自由基聚合反应液的有机溶剂,对所得液体进行柱层析分离。
[0073]
在本发明中,所述去除有机溶剂的方式优选为旋蒸。在本发明中,所述柱层析分离的流动相优选为甲醇和二氯甲烷,所述甲醇和二氯甲烷的体积比优选为1:1。
[0074]
本发明提供了上述活性氧响应型两亲性聚合物作为活性氧靶向的疏水药物递送载体的应用。在本发明中,当聚合物所形成的胶束接触病变部位的活性氧时,腙结构发生断裂,使得聚合物的亲水部分与疏水部分分离,亲水部分会溶解于水中,使得胶束解体。随着胶束的解体,其内包裹的药物可以释放至活性氧水平较高的位置,实现定点治疗。在本发明中,所述病变部位优选为伤口或炎症部位,所述炎症部位优选为肺炎部位或骨关节炎部位。
[0075]
本发明提供了一种载药纳米胶束,包括上述活性氧响应型两亲性聚合物,和被所述活性氧响应型两亲性聚合物分子链段包裹的疏水药物。
[0076]
在本发明中,所述疏水药物优选为姜黄素、阿霉素、富马酸二甲酯和酮洛芬中的一种或几种;在本发明中,所述活性氧响应型两亲性聚合物与疏水药物的质量比优选为10000:1~10,更优选为10000:2~5。
[0077]
在本发明中,所述纳米胶束的粒径优选为6~20nm,更优选为10~15nm。
[0078]
本发明提供了上述载药纳米胶束的制备方法,包括以下步骤:
[0079]
将活性氧响应型两亲性聚合物、疏水药物与醇溶剂混合,去除醇溶剂,得到混合组分;
[0080]
将所述混合组分与水相液体超声混合,得到载药纳米胶束。
[0081]
本发明将活性氧响应型两亲性聚合物、疏水药物与醇溶剂混合,去除醇溶剂,得到混合组分。在本发明中,所述醇溶剂优选为甲醇和/或乙醇。在本发明中,所述混合的方式优选为:
[0082]
将活性氧响应型两亲性聚合物与醇溶剂混合,得到活性氧响应型两亲性聚合物溶液;
[0083]
将疏水药物与醇溶剂混合,得到疏水药物溶液;
[0084]
将活性氧响应型两亲性聚合物溶液与疏水药物溶液混合。
[0085]
在本发明中,所述混合优选为搅拌混合。在本发明中,所述活性氧响应型两亲性聚合物溶液的浓度优选为1~100mg/ml,更优选为5~50mg/ml,更优选为10~30mg/ml;所述疏水药物溶液的浓度优选为0.01mm~1mm,更优选为0.2mm。在本发明中,所述活性氧响应型两亲性聚合物溶液与疏水药物溶液的体积比优选为100;1~1:100,进一步优选为1:5。
[0086]
在本发明中,所述去除有机溶剂的方式优选为旋蒸。
[0087]
本发明将所述混合组分与水相液体超声混合,得到载药纳米胶束。在本发明中,所述水相液体优选为水或pbs缓冲溶液。
[0088]
在本发明中,所述超声混合的功率优选为50~1200w,更优选为200~800w,时间优选为10~120s,更优选为30~60s。
[0089]
所述超声混合后,本发明优选对所得混合液进行过滤,所述过滤优选为使用250μm滤头过滤。
[0090]
下面结合实施例对本发明提供的活性氧响应型两亲性聚合物及其制备方法和作为活性氧靶向的疏水药物递送载体的应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0091]
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0092]
实施例1活性氧响应型两亲性聚合物的制备
[0093]
活性氧响应型两亲性聚合物的合成路线如图1所示,包括以下步骤:
[0094]
(1)4-羟基癸亚胺苯甲酰肼的制备
[0095]
将4-羟基苯甲酰肼、1-癸醛按照摩尔比1:1溶于甲醇中。反应混合体系在65℃油浴下搅拌反应4小时。反应结束后将反应液冷却至室温,溶剂在减压下蒸馏,得到4-羟基癸亚胺苯甲酰肼白色固体(95%)。
[0096]
4-羟基癸亚胺苯甲酰肼的1h-nmr谱图如图2所示。
[0097]
(2)聚合物单体的制备
[0098]
将4-羟基癸亚胺苯甲酰肼、三乙胺按照摩尔比1:1.5溶于四氢呋喃(thf)中。将反应体系在冰浴中冷却至0℃。将摩尔比为1.1甲基丙烯酰氯溶于thf中后,缓慢滴加到反应混合物中。将反应混合物缓慢恢复到温度,然后在室温下搅拌4小时。反应结束后过滤除去沉淀,将溶剂于减压蒸馏下出去。粗产物通过柱层析法(乙酸乙酯/己烷=1:1,v/v)纯化得到聚合物单体(白色固体,78%)。
[0099]
聚合物单体的1h-nmr谱图如图3所示。
[0100]
(3)活性氧响应型两亲性聚合物的制备
[0101]
将聚合物单体与聚乙二醇单甲醚甲基丙烯酸酯(pegma,mn~950g/mol)按照摩尔比1:1溶解于thf中,并加入相对于单体3mol%的偶氮二异丁腈(aibn)作为引发剂。在体系充满氮气后于75℃油浴中反应48小时。将反应液中的thf通过减压蒸馏除去,粗产物通过凝胶柱层析法(甲醇/二氯甲烷=1:1,v/v)纯化得到活性氧响应型两亲性聚合物,记为p1(淡黄色固体,78%)。
[0102]
活性氧响应型两亲性聚合物的1h-nmr谱图如图4所示。
[0103]
实施例2抗氧化纳米胶束的制备
[0104]
(1)姜黄素溶液的制备
[0105]
将一定量的姜黄素加入到甲醇中,搅拌至溶解,得到摩尔浓度为1mm的姜黄素甲醇
溶液。
[0106]
(2)p1溶液的制备
[0107]
将一定量的p1加入甲醇中,得到p1的甲醇溶液,质量浓度为50mg/ml。
[0108]
(3)纳米胶束的制备
[0109]
取1ml姜黄素甲醇溶液与0.1mlp1甲醇溶液,充分混合后减压蒸馏除去溶剂,之后加入10mlpbs溶液,于超声波清洗机中超声30秒,后用250μm滤头过滤,得到包载姜黄素的活性氧响应型纳米胶束。
[0110]
实施例3纳米胶束性质测试
[0111]
1、纳米胶束粒径测试
[0112]
制备纳米胶束样品1ml(室温~25℃),静置约1小时后在室温下用纳米粒径仪测量其胶束的粒径。所得结果如图5所示,可以看出,该胶束的粒径在11nm左右,一般认为该粒径大小的颗粒透过细胞膜的能力较强。
[0113]
连续测试其粒径一星期,所得结果如图6所示,其粒径没有明显的变化,说明该纳米胶束在pbs溶液中稳定存在,不会缓慢分解或聚集。
[0114]
2、临界胶束浓度的测试
[0115]
1、荧光分子氟硼荧(bodipy)溶液的制备
[0116]
将一定量的bodipy分子加入到甲醇中,搅拌至溶解,得到摩尔浓度为0.1mm的bodipy甲醇溶液。
[0117]
2、p1溶液的制备
[0118]
将一定量的p1加入甲醇中,得到p1的甲醇溶液,质量浓度为50mg/ml。之后以该溶液为母液,分别稀释至0.1、1、10、100、400、700、2000μg/ml。
[0119]
3、纳米胶束的制备
[0120]
取50μlbodipy甲醇溶液与0.1ml不同浓度的p1甲醇溶液,充分混合后减压蒸馏除去溶剂,之后加入1mlpbs溶液,于超声波清洗机中超声30秒,后用250μm滤头过滤,得到包载bodipy的不同浓度的纳米胶束。
[0121]
4、纳米胶束荧光强度的测试
[0122]
由于聚合物在形成胶束之前,对疏水性小分子bodipy的包载能力非常弱,而bodipy在水中会聚集导致荧光淬灭,而当胶束形成后,bodipy会被包裹于胶束的疏水性内核中,其荧光会显著增加。因此可以通过该方法测试纳米胶束形成的临界浓度。将上述制备的不同浓度的纳米胶束样品1ml(室温~25℃),静置1小时后用纳全波长微孔板读板机以490nm波长作为激发光,510nm作为发射检测波长,测试各溶液的荧光强度。结果如图7所示,该胶束的临界浓度在0.011mg/ml左右。该临界胶束浓度相较于大多数两亲性聚合物较低,保证了本发明中的聚合物可以在大量稀释的情况下依然保持胶束形态,避免其中的抗氧化物质因为稀释被提前释放。
[0123]
实施例4
[0124]
1、fox标准溶液的配制以及标准曲线的制作
[0125]
(1)配制250mm的硫酸:在烧杯中加入一部分水,加入1.4ml浓硫酸,放热完成后定容成100ml;
[0126]
(2)配制100mm山梨醇:称量9.1085g山梨醇,溶于一部分水中;
[0127]
(3)配制100um二甲酚橙:称量35.831mg二甲酚橙,溶于一部分水;
[0128]
(4)配制250um硫酸亚铁铵:称量49.0175mg硫酸亚铁铵,溶于50ml配制的(1)中,将(2)加入到(4)中,再把(3)加入到(2)和(4)的混合液中,定容到500ml;
[0129]
(5)取200μl fox原液,加入5μl不同浓度的naclo,反应30min时,用紫外测560nm处的吸光度。制作标准曲线,所得结果如图8所示。
[0130]
2、纳米胶束的抗氧化测试
[0131]
向制备好的纳米胶束200μl,加入5μl不同浓度的naclo,搅拌均匀后静置2h,之取出5μl加入到200μlfox工作液中,再次静置30分钟后,用紫外测560nm处的吸光度,其结果如图9所示。由图9可知该材料可以完全消除1.5mm浓度以下的naclo,而炎症细胞以及癌细胞中活性氧的浓度一般不会超过0.1mm,说明该材料可以充分抑制病变细胞中的活性氧。
[0132]
实施例5
[0133]
细胞毒性测试
[0134]
采用cell counting kit-8(cck-8)试剂盒检测p1对小鼠单核巨噬细胞(raw 264.7)的细胞毒性,该细胞是炎症实验常用的细胞。该细胞培养条件为:dmem培养基+15%fbs+1%链霉素-青霉素。稀释细胞原液至5
×
104个细胞/毫升。选用96孔板,每孔加入100μl稀释的细胞悬液置于培养箱培养24小时。使用新鲜培养基配制的不同浓度的p1材料稀释液(浓度分别为0、0.1、0.5、1和5mg/ml),培养24小时后取出孔板,各加入上述配置好的材料稀释液100μl,继续放置在培养箱培养24小时后,使用移液器吸出旧培养基,每孔加入含10%cck-8的基础培养基100μl,孔板置于细胞培养箱避光孵育2小时后,通过酶标仪检测450nm的吸光度值,通过与对照组的对比计算不同浓度下细胞的存活率,所得结果如图10所示。从图10可以看出该材料均有约100%的存活率,表明这种聚合物不具有细胞毒性,是一种安全性较好材料。
[0135]
实施例6
[0136]
药物释放测试
[0137]
向制备好的包覆了5μm荧光分子bodipy的纳米胶束200μl,加入5μl不同浓度的naclo或pbs,搅拌均匀后连续用紫外测其500nm处的吸光度,并将其与初始状态的吸光度作比较,其结果如图11所示。由图11可知该材料在没有naclo刺激下并没有明显的解体以及被包覆物的释放。而随着naclo的加入,纳米胶束有明显的解体,伴随着其中被包覆分子的释放,其中前60分钟的释放最为迅速,之后速度明显放缓。在10mm的naclo下,该胶束可以在2h内释放约80%的被包覆药物。
[0138]
实施例7
[0139]
细胞内ros清除测试
[0140]
培养基稀释c3h细胞至1
×
105个细胞/ml,每孔接种500μl细胞悬液于放置于24孔板中,并分为三组:对照组,naclo组,实验组。培养24h后对照组更换500μl新鲜培养基,其余两组每孔加入500μl用新鲜培养基稀释至1mm的naclo溶液,放置于培养箱继续培养1.5h。之后用pbs洗涤细胞,对照组及naclo组每孔更换500μl新鲜培养液,实验组每孔分别加入500μl用新鲜培养基稀释后的p1+姜黄素溶液,p1浓度为0.5mg/ml。继续孵育12h后,每孔加入ros检测试剂deep red(ab186029),之后加入300μl 4%多聚甲醛固定细胞,再加入dapi染色,洗涤后使用共聚焦显微镜拍照观察统计细胞内的荧光信号如图12所示。对照组的红色荧光
较弱,说明细胞内没有高浓度的ros;naclo组的红色荧光明显增强,说明细胞内部ros含量较高;而实验组也用naclo处理后,由于经过p1+姜黄素处理,其中的ros水平基本恢复到未用naclo处理前的水平,说明本发明中的纳米胶束具有清除细胞内ros的功能。
[0141]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1