一种辛酰溴苯腈的制备方法与流程
1.本发明涉及农药领域,尤其涉及一种辛酰溴苯腈的制备方法。
背景技术:
2.辛酰溴苯腈(分子式:c
15h17
br2no2,分子量:403.11,化学名称:3,5-二溴-4-辛酰氧苯甲腈)是一种广谱、选择性苗后茎叶处理触杀型除草剂。
3.辛酰溴苯腈结构式如下式:
[0004][0005]
辛酰溴苯腈是淡黄色蜡状固体,熔点45-46℃,在90℃/13.33pa下升华。工业品微有油酯气味,在40-45℃以上熔融。不溶于水。溶解度(20-25℃):丙酮、乙醇中100g/l;苯、二甲苯中700g/l;氯仿、二氯甲烷中800g/l;环已酮500g/l。在贮存中稳定,与大多数其它农药不反应,稍有腐蚀性,易被稀碱液水解。对光和熔点下稳定。急性经口ld50:大鼠365mg/kg,小鼠306mg/kg,急性经皮ld50:大鼠>2000mg/kg。喷雾3.4mg辛酰溴苯腈/l,对蜜蜂没有触杀毒性。
[0006]
由于该产品具有显著的除草性能,可用于防除一年生和多年生阔叶杂草,同时,它对禾本科作物具有较高的选择性,能安全使用于作物各个生长期,它广泛用于麦田、玉米、高梁、甘蔗、亚麻、洋葱等多种作物田,防除蓼、藜、苋、麦瓶草、龙葵、苍耳、田旋花等多种阔叶杂草。因此,正越来越多地受到植保部门的关注。
[0007]
法国罗纳普朗克公司(现为拜耳公司)于1987年率先将商品伴地农22.5%乳油在我国进行临时登记(登记号:ls 87026),登记作物为小麦和玉米,防治阔叶杂草。国内企业自1999年由沙隆达郑州农药有限公司首家登记人市(登记号:ls 99546)。
[0008]
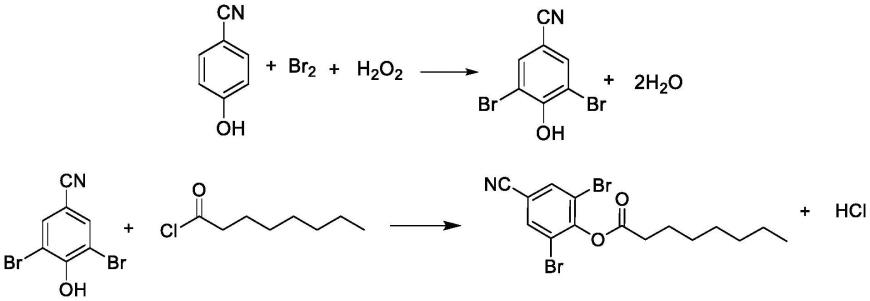
辛酰溴苯腈现有主流路线为对羟基苯甲腈经溴化、酯化得到辛酰溴苯腈。根据溴化、酯化的顺序不同以及所采用的溴化剂不同,有如下合成路线:
[0009]
路线一:以对羟基苯甲腈为原料,先使用溴素、双氧水进行溴化反应生成溴苯腈,再使用辛酰氯酯化得到辛酰溴苯腈。
[0010]
[0011]
例如,专利申请cn108569985a公开了一种高纯辛酰溴苯腈的制备方法,以对氰基酚为原料,使用溴素和双氧水氧化,制得溴苯腈后使用辛酰氯酯化最终制得辛酰溴苯腈。
[0012]
路线二:以对羟基苯甲腈为原料,先使用辛酰氯酯化,再使用溴素、双氧水溴化得到辛酰溴苯腈。
[0013][0014]
例如,专利申请cn104926692a公开的一种辛酰溴苯腈的制备工艺,以对羟基苯甲腈为原料,先完成酯化反应,再完成溴化反应制得辛酰溴苯腈。
[0015]
路线三:与上述路线一相同,但溴化采用的是氢溴酸、双氧水进行,酯化仍采用辛酰氯进行。
[0016][0017]
文献(曹根法,杨建萍,缪维芳.辛酰溴苯腈的合成研究[j].浙江化工,2001(02):50-51.)公开了将溴化过程中溴来源由溴素替换为溴化氢,溴化氢先与双氧水氧化生成溴单质,再与对羟基苯甲腈反应完成溴化。溴化后再用辛酰氯完成酯化反应制得辛酰溴苯腈,采用的是路线三的合成方式。
[0018]
上述合成路线均以对羟基苯甲腈为起始原料,在单步酯化工序中:
[0019][0020]
该步酯化反应,会加入过量的辛酰氯,促进反应的正向进行以保证溴苯腈的转化率,但后续的后处理需要通过碱洗或减压蒸馏的方式除去过量的辛酰氯。
[0021]
碱洗方式中,辛酰氯生成辛酸钠,辛酸钠同时具备七个碳原子的烷烃基团与带负电荷的羧基,其中烷烃基团亲油、羧基亲水,而碱洗过程需要将本就不分层的油水层高转速搅拌混合达到洗涤效果,在混合过程中,该物质起到乳化剂的作用,将两相乳化,分层出现困难。目前生产厂家多采用长时间静置以及离心等物理手段,以达到破乳分层的效果,但该方法仍然存在少量乳化中间层的残留问题,且操作周期长,影响生产效率,并且对设备要求
高,以及带来能耗高、设备投资大和后续设备维修保养费用高等问题。
[0022]
减压蒸馏方式中,考虑到辛酰氯沸点为195℃,需要加热至110-130℃,且对真空度要求高,能耗高,后续还需降温装桶,频繁的升温降温造成大量热损失,成本较高,生产周期较长,而且该方法也无法将其中的辛酰氯完全除净。
[0023]
因此,如何去除辛酰氯是亟待解决的技术问题。
技术实现要素:
[0024]
发明目的
[0025]
为克服上述不足,本发明的目的在于提供一种辛酰溴苯腈的制备方法,该方法可以避免辛酰溴苯腈酯化反应完成后水洗碱洗后处理过程中出现的乳化现象,进而减少后续乳化层的处理装置,同时也可以避免现有技术中需在110~130℃减压至-0.095mpa以去除多余辛酰氯的高温减压蒸馏过程。
[0026]
解决方案
[0027]
为实现本发明目的,本发明采用的技术方案如下:
[0028]
第一方面,本发明提供了一种辛酰溴苯腈的制备方法,将含有辛酰氯和辛酰溴苯腈的反应液中,在洗涤时加入氨基糖类,洗涤搅拌,静置分层,获得含有辛酰溴苯腈的油相。
[0029]
进一步地,所述氨基糖类包括葡糖胺、半乳糖胺、盐酸氨糖、硫酸氨糖、单糖胺类中的一种或几种;可选地,所述单糖胺类包括果糖胺、木糖胺、核糖胺、甘露糖胺、岩藻糖胺、来苏糖基胺、阿拉伯糖胺中的一种或几种;可选地,所述氨基糖类为葡糖胺。
[0030]
进一步地,氨基糖类在体系中的质量分数为0.1%~8%,可选地为0.1~3%,可选地为0.1~1.5%。
[0031]
进一步地,所述含有辛酰氯和辛酰溴苯腈的反应液,为辛酰氯和溴苯腈反应获得的反应液。
[0032]
进一步地,所述含有辛酰氯和辛酰溴苯腈的反应液为辛酰氯和溴苯腈在无溶剂时进行反应的反应液,洗涤方式为碱洗。
[0033]
进一步地,辛酰氯和溴苯腈的反应温度100-140℃;可选地,碱洗采用碱金属氢氧化物溶液;可选地,碱金属氢氧化物选自氢氧化钠和氢氧化钾中的一种或两种;可选地,碱洗液中含有1~10%w/w的碱,可选地含有2~5%的碱;可选地,碱洗的温度为80~90℃;可选地,碱洗液中氨基糖类的浓度为0.1~8%,可选地为0.5~5%。
[0034]
进一步地,所述含有辛酰氯和辛酰溴苯腈的反应液为辛酰氯和溴苯腈在溶剂中进行反应时的反应液,洗涤方式为水洗。水洗液中氨基糖类的浓度为0.1~8%,可选地为0.5~5%;
[0035]
和/或,溶剂选自沸点大于105℃的溶剂,可选地溶剂为甲苯、二甲苯、氯苯;可选地,溶剂中含有碱,可选地,辛酰氯与碱的摩尔比为1:1~1.15,可选地,碱选自碱金属氢氧化物溶液或三乙胺;可选地溶剂中含有氢氧化钠、氢氧化钾和/或三乙胺;可选地,溶剂中含有10~50%w/w的碱,可选地,溶剂中含有20~50%w/w的碱。
[0036]
进一步地,所述氨基糖类物质:溴苯腈的质量配比为1:500-1:20。
[0037]
进一步地,所述氨基糖类物质:溴苯腈的质量配比为1:200-1:30,可选地为1:150-1:50。
[0038]
进一步地,油相的处理方式为:减压蒸馏回收溶剂,油相降温获得辛酰溴苯腈;可选地减压蒸馏回收溶剂的条件为:50-60℃,0.06-0.09mpa。
[0039]
有益效果
[0040]
(1)本发明的制备方法在后处理过程中引入极少量氨基糖类物质,可以避免辛酰溴苯腈酯化反应完成后水洗碱洗后处理过程中出现的乳化现象,进而减少后续乳化层的处理装置,同时也可以避免高温减压蒸馏过程。且氨基糖类物质与辛酸成盐后溶解于水相中,对原反应收率、产品品质无任何影响;
[0041]
(2)本发明的方法适用性广,在有无溶剂的酯化反应后处理中,均可使用。
[0042]
(3)本发明的后处理方法工艺稳定,重复性强,成本低,可以进行大吨位连续化生产。
具体实施方式
[0043]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0044]
另外,为了更好的说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在一些实施例中,对于本领域技术人员熟知的原料、方案、方法、手段等未作详细描述,以便于凸显本发明的主旨。
[0045]
除非另有其它明确表示,否则在整个说明书和权利要求书中,术语“包括”或其变换如“包含”或“包括有”等等将被理解为包括所陈述的元件或组成部分,而并未排除其它元件或其它组成部分。
[0046]
以下实施例中的产物含量通过液相或气相色谱仪确认,反应过程中,纯度和收率采用外标法计算得到。
[0047]
以下实施例中的溴苯腈、辛酰氯等原料可市售获得。
[0048]
溴苯腈与辛酰氯酯化反应生成辛酰溴苯腈的路线:
[0049][0050]
该反应路线中,有三种常规路线:
[0051]
1、酯化反应无溶剂法
[0052]
使用溴苯腈与辛酰氯在100-140℃高温下,保温6-10h(或使用高真空-0.095mpa,保温4-5h),合格后使用高真空-0.09mpa,蒸馏过量的辛酰氯,合格后油相降温至60-80℃装桶降温。(或合格后直接降温至80-90℃,进行一次稀碱液洗涤,长时间需要30-60min静置分层,待不再分层后,分出乳化层单独回收产品,油相装桶降温)。
[0053]
2、酯化溶剂法
[0054]
使用溴苯腈、辛酰氯与甲苯等高沸点溶剂、氢氧化钠溶液或三乙胺,在100-140℃
高温下,保温6-12h,合格后加入水洗涤,长时间需要30-90min静置分层,待不分层后,分出乳化层单独处理回收,再使用减压蒸馏回收溶剂,油相降温装桶;
[0055]
上述两种合成方法一般还会回收乳化层,乳化层的回收处理方法一般为:
[0056]
取乳化层升温至30-60℃溶解于甲醇等有机醇类、烷烃类、芳香类溶剂中,保温搅拌0.5-2h,降温至0-5℃,抽滤降温得到回收产品,抽滤的回收溶剂多次套用后,蒸馏回收,蒸馏釜残做危废处理。
[0057]
上述两种合成方法中,碱液洗涤后静置分层需要较长时间,本发明在碱液或水洗涤时加入极少量的氨基糖类,过量的辛酰氯水解后生成的辛酸与氨基糖类物质反应后生成溶于水相中的物质,可以实现快速分层,一般3~10min就能完成分层,避免了乳化层的出现,解决了后续乳化难分层的难题,减少了设备投资和运行成本,节约了能耗,再减压蒸馏回收溶剂,可以大大提高生产效率。
[0058]
实施例1
[0059]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温130℃保温6h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g、氨基葡萄糖0.8g搅拌洗涤15min,保持80℃分液静置2min得到水相115.71g、油相124.44g,装桶降温得到辛酰溴苯腈124.44g,产品纯度98%,收率97.6%。
[0060]
实施例2
[0061]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温130℃保温6h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g、岩藻糖胺0.8g搅拌洗涤10min,保持80℃分液静置10min得到水相117.04g、油相123.81g,装桶降温得到辛酰溴苯腈123.81g,产品纯度97.7%,收率96.8%。
[0062]
实施例3
[0063]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温130℃保温6h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g、阿拉伯糖胺0.8g搅拌洗涤20min,保持80℃分液静置10min得到水相115.92g、油相124.71g,装桶降温得到辛酰溴苯腈124.71g,产品纯度97.3%,收率97.1%。
[0064]
实施例4
[0065]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、三乙胺34.24g(0.335mol)、甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入水100g、氨基葡萄糖1g搅拌洗涤15min,保持80℃静置3min分液得到水相151.34g、油相323.54g,减压蒸馏回收溶剂甲苯198.2g,蒸馏完毕后装桶降温得到辛酰溴苯腈123.81g,产品纯度98.1%,收率97.2%。
[0066]
实施例5
[0067]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、三乙胺34.24g(0.335mol)、甲苯200g,保持体系内温110℃滴加1h辛酰氯
55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入水100g、来苏糖基胺1g搅拌洗涤15min,保持80℃静置11min分液得到水相150.1g、油相324.94g,减压蒸馏回收溶剂甲苯197.86g,蒸馏完毕后装桶降温得到辛酰溴苯腈125.74g,产品纯度96.9%,收率97.5%。
[0068]
实施例6
[0069]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温100℃、负压0.095mpa保温4h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g、半乳糖胺0.6g搅拌洗涤15min,保持80℃静置5min分液,得到水相116.21g、油相124.71g,油相装桶降温得到辛酰溴苯腈124.71g,产品纯度97.3%,收率97.1%。
[0070]
实施例7:
[0071]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温100℃、负压0.095mpa保温4h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g、核糖胺0.6g搅拌洗涤15min,保持80℃静置3min分液,得到水相115.88g、油相124.83g,油相装桶降温得到辛酰溴苯腈124.83g,产品纯度96.9%,收率96.8%。
[0072]
实施例8
[0073]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、50%氢氧化钠溶液26.8g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入50g水、果糖胺0.7g搅拌洗涤15min,保持80℃静置10min分液得到水相96.86g、油相321.12g,减压蒸馏回收溶剂二甲苯197.5g,蒸馏完毕后装桶降温得到辛酰溴苯腈122.89g,产品纯度96.7%,收率95.1%。
[0074]
实施例9
[0075]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、50%氢氧化钠溶液26.8g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入50g水、木糖胺0.7g搅拌洗涤15min,保持80℃静置14min分液得到水相96.43g、油相320.87g,减压蒸馏回收溶剂二甲苯197.34g,蒸馏完毕后装桶降温得到辛酰溴苯腈122.13g,产品纯度96.9%,收率94.7%。
[0076]
实施例10
[0077]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、50%氢氧化钠溶液26.8g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入50g水、盐酸氨糖0.7g搅拌洗涤15min,保持80℃静置25min分液得到水相95.96g、油相321.32g,减压蒸馏回收溶剂二甲苯198.61g,蒸馏完毕后装桶降温得到辛酰溴苯腈121.24g,产品纯度97.4%,收率94.5%。
[0078]
本实施例中采用盐酸氨糖需要静置的时间略长,可能与盐酸氨糖含有氯化氢有关系,但在静置过程中不会产生乳化层,相对现有技术也具有明显的提高。
[0079]
实施例11
[0080]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、20%氢氧化钾溶液93.98g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温7h,保温结束降温并保持在80℃,加入50g水、葡糖胺1.6g搅拌洗涤15min,保持80℃静置5min分液得到水相165.54g、油相320.49g,减压蒸馏回收溶剂二甲苯197.46g,蒸馏完毕后装桶降温得到辛酰溴苯腈121.92g,产品纯度97.3%,收率94.9%。
[0081]
实施例12
[0082]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温100℃、负压0.095mpa保温4h,保温结束降温并保持在80℃,加入5%氢氧化钾溶液100g、甘露糖胺1.34g搅拌洗涤15min,保持80℃静置10min分液,得到水相116.71g、油相125.23g,油相装桶降温得到辛酰溴苯腈125.23g,产品纯度96.7%,收率96.9%。
[0083]
对比例1
[0084]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温130℃保温6h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g搅拌洗涤15min,保持80℃分液静置10min,乳化层较厚,获得水相108.2g、油相112.1g、乳化中间层20.3g,继续静置至40min才能得到水相115.2g、油相122.4g、乳化中间层3g,油相装桶降温得到辛酰溴苯腈122.4g,产品纯度98%,收率96%。
[0085]
将3g乳化中间层加入30g甲醇中,升温至30℃,保温30min,结束后降温至5℃,抽滤得到回收辛酰溴苯腈1.67g,产品纯度97.4%,收率与前一步合并为97.3%。
[0086]
对比例2
[0087]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、三乙胺34.24g(0.335mol)、甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入水100g搅拌洗涤15min,保持80℃分液静置,得到水相130.08g、油相297.74g、乳化中间层45.3g,继续保温静置至45min才能得到水相141.42g、油相315.63g、乳化中间层16.07g,减压蒸馏回收溶剂甲苯190.2g,蒸馏完毕后装桶降温得到辛酰溴苯腈123.93g,产品纯度97.5%,收率96.7%。
[0088]
将16.07g乳化中间层加入40g乙醇中,升温至40℃,保温60min,结束后降温至5℃,抽滤得到回收辛酰溴苯腈1.29g,产品纯度97.2%,收率与前一步合并为97.7%。
[0089]
对比例3
[0090]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的500ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、辛酰氯55.01g(0.335mol),保持体系内温100℃、负压0.095mpa保温4h,保温结束降温并保持在80℃,加入2%氢氧化钠溶液100g搅拌洗涤15min,保持80℃分液静置至少60min才能分液得到水相112.2g、油相122.51g、乳化中间层5.3g,油相装桶降温得到辛酰溴苯腈122.51g,产品纯度97.1%,收率95.2%。
[0091]
将5.3g乳化中间层加入30g二氯乙烷中,升温至30℃,保温30min,结束后降温至5
℃,抽滤得到回收辛酰溴苯腈1.53g,产品纯度97.8%,收率与前一步合并为96.4%。
[0092]
对比例4
[0093]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、50%氢氧化钠溶液26.8g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温5h,保温结束降温并保持在80℃,加入水50g搅拌洗涤15min,保持80℃分液静置至少50min才能得到水相87.36g、油310.66g、乳化中间层19.68g,减压蒸馏回收溶剂二甲苯188.12g,蒸馏完毕后装桶降温得到辛酰溴苯腈121.36g,产品纯度97.1%,收率94.3%。其中,碱溶液为氢氧化钠溶液。
[0094]
将19.68g乳化中间层加入60g二甲苯中,升温至40℃,保温60min,结束后降温至5℃,抽滤得到回收辛酰溴苯腈0.77g,产品纯度96.9%,收率与前一步合并为94.9%。
[0095]
对比例5
[0096]
在装有机械搅拌装置、油浴、温度计和滴液漏斗的1000ml四口烧瓶内,加入溴苯腈86.71g(0.31mol)、20%氢氧化钾溶液93.98g(0.335mol)、二甲苯200g,保持体系内温110℃滴加1h辛酰氯55.01g(0.335mol),滴加完毕后保持温度保温6h,保温结束降温并保持在80℃,加入水50g搅拌洗涤15min,保持80℃分液静置至少50min才能得到水相154.28g、油308.68g、乳化中间层21.86g,减压蒸馏回收溶剂二甲苯185.33g,蒸馏完毕后装桶降温得到辛酰溴苯腈122.18g,产品纯度96.5%,收率94.3%。
[0097]
将21.86g乳化中间层加入60g二甲苯中,升温至40℃,保温60min,结束后降温至5℃,抽滤得到回收辛酰溴苯腈1.26g,产品纯度97.8%,收率与前一步合并为95.3%。
[0098]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1