人乳头瘤病毒分型及相关基因甲基化一体化检测模型及其构建方法与流程
1.本发明涉及分子生物学分析检测技术领域,具体涉及一种人乳头瘤病毒分型及相关基因甲基化一体化检测模型及其构建方法。
背景技术:
2.目前宫颈癌的筛查包括宫颈细胞学检测及人乳头瘤病毒(hpv)检测。细胞学检测包括巴氏涂片和tct液基细胞压片,其诊断水平受医师主观因素影响较大。hpv dna检测相比细胞学检测,能够对高危型和低危型进行分型和定量检测,但是大部分hpv为一过性感染,不会发展为宫颈癌前病变或宫颈癌,但易给妇女造成不必要的心理负担。因此单一依靠hpv筛查,会造成过度诊断和过度治疗。
技术实现要素:
3.本发明旨在提供一种人乳头瘤病毒分型及相关基因甲基化一体化检测模型及其构建方法以弥补上述技术的不足。本发明采用了如下的技术方案:基因甲基化是指dna分子上cpg双核苷中的胞嘧啶(c)在酶的作用下选择性地添加甲基形成5'-甲基胞嘧啶的过程。cpg双核苷常位于基因转录调控区附近,其甲基化能引起染色质结构、dna构象、dna稳定性等发生改变,从而调控基因的转录和表达。基因甲基化异常是肿瘤发生最常见的表观遗传变化之一,肿瘤的发生表现为基因组整体甲基化水平降低(癌基因)和cpg岛局部甲基化水平的异常升高(抑癌基因)。因此通过检测hpv基因组甲基化及人宫颈癌相关基因甲基化可以对具有潜在病变进展风险的患者进行分流。
4.本发明提供一种人乳头瘤病毒分型及相关基因甲基化一体化检测模型及其构建方法,包括:提取待测宫颈细胞dna,将所述dna片段化,进行末端修复和接头连接;对上述产物进行甲基化处理,再进行pcr富集和质检;根据宫颈癌相关基因和18种hpv基因组的全长序列,设计探针;所述18种hpv基因组包括hpv6、hpv11、hpv16、hpv18、hpv31、hpv33、hpv35、hpv39、hpv45、hpv51、hpv52、hpv56、hpv58、hpv59、hpv66、hpv68a、hpv69和hpv82;通过所述探针靶向富集目标区域的核酸,进行高通量测序;建立人乳头瘤病毒分型及相关基因甲基化一体化检测模型,进行生信分析,比对hpv分型;从序列与18种hpv型别比对的结果中,分别提取分型各基因所包含的reads,计算基因甲基化分值,并统计基因覆盖深度,以甲基化分值与基因覆盖深度作为hpv基因甲基化高低程度指标,对样本进行分类预测;对宫颈癌相关基因甲基化进行检测。
5.进一步,所述比对hpv分型包括:使用bismark将预处理后数据与18种hpv型别的参
考基因组进行比对,得到序列比对结果的详细信息,计算得到覆盖度,以覆盖度大于40%筛选数据得到基本分型信息。
6.进一步,所述宫颈癌相关基因包括:adcyap1、ajap1、ankrd18cp、apc、astn1、cadm1、ccna2、cdh1、cdh13、cdh6、cdkn2a、dapk1、dlx1、epb41l3、fam19a4、fanci、fhit、gata4、gfra1、hs3st2、jam3、kcnip4、lhx8、lmx1a、mal、mir124-1、mir124-2、mir124-3、pax1、pcdha13、pcdha4、pou4f3、rarb、rnaseh2a、rubcnl、rxfp3、slit2、sox1、sox17、st6galnac5、tert、timp3、wif1、znf671和zscan1。
7.进一步,所述设计探针包括:获取所述宫颈癌相关基因及所述18种hpv基因组全长序列的甲基化区域,提取参考序列,根据参考序列的正向序列和反向序列,设计经过亚硫酸盐处理后的模拟高甲基化和低甲基化序列,然后以这些序列为模板从第一个碱基开始截取120bp的序列做探针,再一次往后移动 n 个碱基,截取120bp的序列做探针,直到最后一个120bp。
8.所述以甲基化分值与基因覆盖深度作为hpv基因甲基化高低程度指标,对样本进行分类预测包括:将每种hpv分型的每个基因区域的甲基化分值与基因覆盖深度相乘,得到一个判断值,若所述判断值大于等于1.04,则判定为cin2+;若所述判断值小于1.04,则判定为≤cin1。
9.进一步,所述计算基因甲基化分值通过下式进行计算:其中,mhl为甲基化分值,l为宫颈癌相关基因含有的cpg位点,mhi为i个连续cpg位点完全甲基化的比例。
10.进一步,所述甲基化分值与宫颈病变程度呈正相关。
11.进一步,所述对宫颈癌相关基因甲基化进行检测包括:比对至人宫颈癌相关基因组生成bam文件,去冗余提取所有cpg位点,再从所述所有cpg位点中提取宫颈癌相关基因cpg位点,分别提取各高密度高关联区域所包含的reads,计算区域内甲基化分值,以甲基化分值作为宫颈癌相关基因甲基化高低程度指标,对样本进行分类预测。
12.另一方面,本发明提供一种人乳头瘤病毒分型及甲基化一体化检测模型,包括:宫颈细胞dna提取模块,用于提取待测宫颈细胞dna,将所述dna片段化,进行末端修复和接头连接;测序模块,用于对上述产物进行甲基化处理,再进行pcr富集和质检;探针设计模块,用于根据宫颈癌相关基因和18种hpv基因组全长序列,设计探针;所述18种hpv基因组包括hpv6、hpv11、hpv16、hpv18、hpv31、hpv33、hpv35、hpv39、hpv45、hpv51、hpv52、hpv56、hpv58、hpv59、hpv66、hpv68a、hpv69和hpv82;富集测序模块,用于通过所述探针靶向富集目标区域的核酸,进行高通量测序;生信分析模块,用于进行生信分析,比对hpv分型;再比对至人宫颈癌相关基因组生成bam文件,去冗余提取所有cpg位点,再从所述所有cpg位点中提取宫颈癌相关基因cpg位点,计算每种hpv分型的每个基因区域的甲基化
分值,以每种hpv分型的每个基因区域的甲基化分值与基因覆盖深度作为hpv基因甲基化高低程度指标,对样本进行分类预测;对宫颈癌相关基因甲基化进行检测。
13.进一步,所述比对hpv分型包括使用bismark将预处理后数据与18种hpv参考基因组序列进行比对,得到序列比对结果的详细信息,计算得到覆盖度,以覆盖度大于40%筛选数据得到基本分型信息;所述以甲基化分值与基因覆盖深度作为hpv基因甲基化高低程度指标,对样本进行分类预测包括:将每种hpv分型的每个基因区域的甲基化分值与基因覆盖深度相乘,得到一个判断值,若所述判断值大于等于1.04,则判定为cin2+;若所述判断值小于1.04,则判定为≤cin1。
14.进一步,所述对宫颈癌相关基因甲基化进行检测包括:比对至人宫颈癌相关基因组生成bam文件,去冗余提取所有cpg位点,再从所述所有cpg位点中提取宫颈癌相关基因cpg位点,分别提取各高密度高关联区域所包含的reads,计算区域内甲基化分值,以甲基化分值作为宫颈癌相关基因甲基化高低程度指标,对样本进行分类预测。
15.本发明的有益效果在于:本发明通过靶向测序的方法可以同时检测hpv分型和评估hpv甲基化及人宫颈癌相关基因甲基化程度,捕获效率高、捕获稳定性和均一性较好,减少测序成本,避免单一hpv检测的过度诊断问题。
附图说明
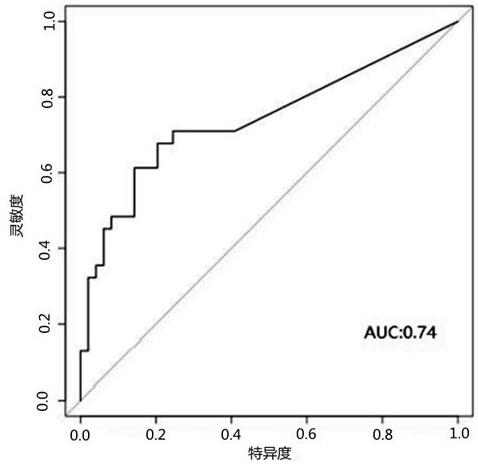
16.图1为实施例3hpv甲基化auc值图;图2为实施例3mhl值与宫颈病变程度关系图。
具体实施方式
17.下面结合具体实施例对本发明作进一步的详细说明。
18.需要说明的是,这些实施例仅用于说明本发明,而不是对本发明的限制,在本发明的构思前提下本方法的简单改进,都属于本发明要求保护的范围。
19.实施例1hpv基因组分型/甲基化及人宫颈癌相关基因组甲基化检测方法建立1、探针设计(1)人宫颈癌相关基因组甲基化mark基因筛选筛选宫颈癌相关的甲基化特征明显的甲基化区域,取每个区域的参考序列(参考基因组版本hg19),去掉重复区域的序列,重复序列采用repeatmask软件分析得到基因序列,参见表1。
20.表1基因列表adcyap1cdh6gfra1mir124-3slit2ajap1cdkn2ahs3st2pax1sox1ankrd18cpdapk1jam3pcdha13sox17apcdlx1kcnip4pcdha4st6galnac5astn1epb41l3lhx8pou4f3tert
cadm1fam19a4lmx1ararbtimp3ccna2fancimalrnaseh2awif1cdh1fhitmir124-1rubcnlznf671cdh13gata4mir124-2rxfp3zscan1(2)探针设计从上述45个基因以及提取的hpv 6、11、16、18、31、33、35、39、45、51、52、56、58、59、66、68a、69和82等18种hpv基因组全长序列的甲基化区域的cpg位点及前后75bp的位置,提取参考序列(参考基因组版本hg19)根据参考序列的正向序列和反向序列,设计经过亚硫酸盐处理后的模拟高甲基化和低甲基化序列,然后以这些序列为模板从第一个碱基开始截取120bp的序列做探针,再一次往后移动 n 个碱基,截取120bp的序列做探针,直到最后一个120bp。每个区域根据外显子的gc含量不同,n会有变化,gc含量太高或太低n越小,探针设计越密集捕获达到的均一性越高。
21.2、宫颈细胞dna提取(dna提取)采用以下任一方式提取宫颈细胞dna:宫颈脱落细胞dna提取:取300μl保存液保存细胞按照通用型柱式基因组提取试剂盒(康为,cwy004)说明书提取基因组dna后qubit检测浓度。
22.石蜡组织或石蜡切片基因组dna提取:取10μm厚的石蜡块或8-10片石蜡切片样本按照generead dna ffpe kit(qiagen,180134)说明书提取基因组dna后qubit检测浓度。
23.新鲜组织基因组dna提取:取25 mg新鲜组织按照通用型柱式基因组提取试剂盒(康为,cwy004)说明书提取基因组dna后qubit检测浓度。
24.3、基因组dna甲基化文库构建(1)甲基化防污染接头设计及合成甲基化防污染接头由通用序列、防污染标签两部分构成,防污染标签包含8-10碱基(随机序列),且接头中的胞嘧啶(c)碱基全部进行甲基化修饰,将合成的接头的干粉用高速离心机12000 rpm离心,加入一定体积的ph为8.0、10mm tris-hcl稀释至100μm;将接头1和接头2各取50μl 100μm退火后使用。
25.(2)基因组dna甲基化文库构建a.基因组dna片段化使用covaris超声打断仪按照相关的说明书将从细胞或组织中获得dna片段化至200bp。
26.将步骤2中组织或细胞dna利用covaris超声打断仪(covaris,s220)按照参数peak incident power 175w、duty factor 10%、cycles per burst 200、treatment time 180s片段化至200bp左右。
27.b.末端修复、连接接头按照2000:1的比例在片段化dna中加入lambda dna后使用rapid max dna lib prep kit for illumina(abclonal,rk20217)试剂盒进行末端修复、连接接头。其中接头为步骤(1)中合成的甲基化修饰接头。
28.c.文库甲基化处理使用epitect plus dna plus dna bisufite kit(qiagen,59124)甲基化处理试
剂盒按照说明书对上一步产物进行甲基化处理。
29.d.pcr富集将上一步甲基化产物使用kapahifi高保真甲基化文库扩增试剂(kapa,kk2802)进行扩增,使用magbeaddnapurificationkit(康为,cw2508m)试剂盒纯化。
30.e.文库质检上一步产物用qubit检测浓度。
31.4、文库靶向富集将待测样本细胞或组织基因组dna为文库,以设计的探针,按照专利201810580442.6实施例1的试剂和实施例2的方法进行靶向捕获。
32.5、生信分析将步骤4得到的靶序列捕获文库通过nextseq500、xten、novaseq等二代测序平台进行高通量测序,得到测序原始数据进行以下分析。
33.(1)原始数据质控a.碱基识别使用illumina官方软件bcf2fastq(version2.15.0.4),根据样本index序列,将illumina测序仪下机二进制bcf格式文件转化并拆分为单个样本可读文件fastq格式。
34.b.数据质控使用cutadapt(version1.16)去除测序接头,删除低质量碱基,生成cleanreads。其中cutadapt(version1.16)的参数为(-q10,10
‑‑
nextseq-trim=10-aatctcgtatgccgtcttctgcttg-aagatcggaagagcgtcgtgtagggaaagagtgtagatctcggtggtcgccgtatcatt),序列长度小于80。
35.(2)hpv分型分析使用bismark将预处理后数据与18种hpv型别的参考基因组进行比对(-n1-p6-l30-most_valid_alignments3),得到序列比对结果的详细信息(bam文件)。将bam中的信息转成bed文件计算得到覆盖度统计文件(sample.depth.coverage),以覆盖度大于40%筛选数据得到基本分型信息。
36.(3)数据比对使用甲基化比对专用软件bismark(v0.17.0)将cleanreads比对至人宫颈癌相关基因组hg19及hpv基因组,其中bismark比对参数为(-un
‑‑
genome_folder-n1-p8-l30-most_valid_alignments3-b
‑‑
samtools_path
–o‑‑
path_to_bowtie-1-2),生成bam文件。
37.使用bismark中的deduplicate_bismark模块对对比后的bam文件去冗余。
38.使用bismark中的bismark_methylation_extractor模块对去冗余的bam文件提取cpg位点信息,参数为(-p
‑‑
no_overlap
‑‑
ignore4
‑‑
ignore_r24
‑‑
samtools_path
‑‑
bedgraph
‑‑
buffer_size20g
‑‑
cytosine_report
‑‑
genome_folder-o./
‑‑
multicore10bam)。
39.使用bismark将预处理后数据与18种hpv型别的参考基因组进行比对,得到序列比对结果的详细信息,计算得到覆盖度,以覆盖度大于40%筛选数据得到基本分型信息;(4)cpg位点提取
bismark_methylation_extractor模块提取的是所有的cpg位点信息,目标基因是宫颈癌相关基因的cpg位点,因此需要从所有的cpg位点信息中提取目标cpg位点信息。使用编写的s_extract_from_bed.pypython脚本从所有的cpg位点信息中提取目标cpg位点信息。
40.(5)对每种分型的每个基因区域计算一个甲基化分值。
41.甲基化分值计算公式为其中l为hpv基因cpg位点,mhi为i个连续cpg位点完全甲基化的比例,p为i个连续cpg位点的片段中,完全甲基片段占比。
42.(6)甲基化程度预测分析从序列与18种hpv型别比对的结果中,分别提取分型各基因所包含的reads,计算基因甲基化分值(mhl),并统计基因覆盖深度,以mhl值与基因覆盖深度作为hpv基因甲基化高低程度指标,对样本进行分类预测;比对至人宫颈癌相关基因组生成bam文件,去冗余提取所有cpg位点,再从所述所有cpg位点中提取宫颈癌相关基因cpg位点,分别提取各高密度高关联区域所包含的reads,计算区域内甲基化分值(mhl),以mhl值宫颈癌基因甲基化高低程度指标,对样本进行分类预测。
43.人宫颈癌相关基因甲基化检测靶标是探针设计区域里的cpg位点,包括:cpg高密度区域划分,即区域内所有相连的cpg位点间距小于100bp;对cpg高密度区域筛选高关联度位点集;两位点同时甲基化或同时非甲基化认为两位点具甲基化关联性。两位点甲基化关联度定义为:两位点同时甲基化或同时非甲基化支持序列数占两位总覆盖序列的比例。高关联度位点集定义为,位点集内任意一个位点与其余至少一个位点关联度大于等于0.8。通过此规则进一步划分位点分组(称位点集),过滤干扰位点;对每一个位点集计算一个甲基化分值甲基化分值计算公式为其中l为宫颈癌相关基因含有的cpg位点,mhi为i个连续cpg位点完全甲基化的比例,p为i个连续cpg位点的片段中,完全甲基片段占比,为了更有效地区分宫颈炎/cin1与cin2/cin3/宫颈癌,计算甲基化分值的时候i从4开始。
44.实施例2hpv分型检测经患者知情同意,采集6例待检患者宫颈脱落细胞样本,用实施例1的方法检测hpv型别,结果见表2:结果中第一列为样本编号;第二列为病毒参考id;第三列为平均测序深度;第四列为目标区域上的覆盖reads数;第五列为目标区域上的覆盖度;第六列为目标区域碱基测序深度不低于20x所占的比例;第七列为测序的reads覆盖到hpv的型别及对应的基因。从数据可以看到,本方法可以对hpv基因组有较好的覆盖,平均深度可达到上万x(20x覆盖可达到100%),可以覆盖到常见hpv基因型,且与荧光定量检测结果一致。
45.表2 6例待检患者宫颈脱落细胞样本检测结果
实施例3hpv甲基化检测hpv基因甲基化水平可通过参与病毒基因与宿主细胞整合过程,并调控癌基因表达、病毒增殖和机体免疫逃逸,促使子宫颈癌发生。本发明在患者知情同意的情况下,使用实施例1的试剂盒和方法对80例hpv感染伴有不同病变的宫颈脱落细胞样本(≤cin1及cin2
+)检测(检测结果见表3),通过提取hpv基因组的cpg位点、对每一个位点集计算一个甲基化值(mhl),并将mhl值与基因区域平均深度相乘,得到一个判断值,并通过youden'sjstatistic算法确定阈值为1.04(灵敏度为79.59%、特异度为67.74%、auc值为0.74)(图1),且判断值与宫颈病变程度呈正相关,若判断值大于等于1.04,则判定为cin2+;若判断值小于1.04,则判定为≤cin1(图2)。
46.表380例hpv感染伴有不同病变的宫颈脱落细胞样本检测结果实施例4hpv分型、hpvdna甲基化和人宫颈癌相关基因甲基化一体化检测经患者同意,取50例待检的宫颈脱落细胞用实施例1的方法进行hpv分型、hpvdna甲基化和人宫颈癌相关基因甲基化的检测,结果见表4:第二列为hpv分型结果;第三列和第四列为hpvdna甲基化检测结果;第五列为人宫颈癌相关基因组dna甲基化判断结果;第六列为组织病理结果。
47.表450例待检的宫颈脱落细胞一体化检测结果
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管通过参照本发明的优选实施例已经对本发明进行了描述,但本领域的普通技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离所附权利要求书所限定的本发明的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1