Paxlovid和波普瑞韦中间体的合成方法与流程
paxlovid和波普瑞韦中间体的合成方法
技术领域
1.本发明属于医药和精细化工领域。具体地说,本发明涉及(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐的新合成方法。
背景技术:
[0002]
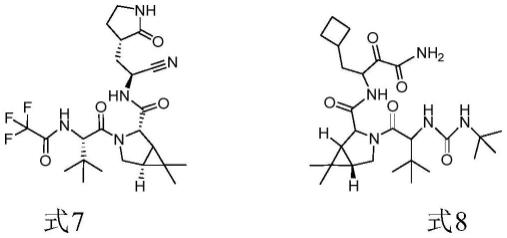
paxlovid是美国辉瑞公司研制的3cl蛋白酶抑制剂,于2021年12月获美国fda批准上市用于抗新冠病毒。paxlovid的主要活性成分之一是为pf-07321332(式7化合物)。
[0003]
波普瑞韦(式8化合物)由美国先灵公司研发,于2011年上市,用于治疗慢性丙型肝炎。
[0004][0005]
式1化合物是合成pf-07321332和波普瑞韦的共同关键中间体,其化学名为:(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐。化学结构如下所示:
[0006][0007]
现有技术中对于式1化合物的合成有报道。例如,中国专利申请cn202111547312.0(申请日:2021-12-16)报道了式1的合成方法,具体合成路线如下所示:
[0008][0009]
该方法以反式-4-羟基-l-脯氨酸衍生物(式2)经过烷基或芳基磺酰氯反应制得式3化合物,式3化合物发生消除反应得式4化合物,式4化合物与磷叶立德试剂反应制得式6化合物,式6化合物脱保护成盐制得式1化合物。第一步磺化反应和第二步消除反应制得式4化合物的收率72%~83%,第三步式4和磷叶立德试剂反应生成式6化合物的收率90%~92%,最后一步脱保护成盐制得式1化合物收率82%~91%。此发明的反应步骤较短,收率较高。但在制备磷叶立德试剂时用到了危险的自燃试剂,正丁基锂,生产危险性大。
[0010]
中国专利申请cn202210054245.7(申请日:2022-1-28)报道了式1化合物的合成方法,该法使用危险的试剂2-重氮丙烷引入异丙基。该法还使用2,2-二溴丙烷试剂引入异丙基,2,2-二溴丙烷试剂比2-二溴丙烷多一个溴,并且2,2-二溴丙烷试剂比2-二溴丙烷试剂价格贵,因此该方法的原子经济性不高。反应也用到危险的可自燃试剂,正丁基锂或锌铜金属试剂催化反应,生产危险性还是很大。
[0011]
中国专利申请cn202111547312.0(申请日:2022-5-56)报道了式1化合物的合成方法,该法使用2,2-二溴丙烷试剂引入异丙基,2,2-二溴丙烷试剂比2-二溴丙烷多一个溴,2,2-二溴丙烷试剂比2-二溴丙烷试剂价格贵,因此原子经济性不高。该反应用到二溴化钴和配体催化反应,因此存在配体制备复杂,二溴化钴价格贵的缺陷。该反应还用到锌粉,锌粉固液两相反应,对生产设备和搅拌方式的要求较高。
[0012]
因此,本领域急需合成(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐的新方法。这种方法应该具备操作简便、安全,对环境和操作人员友好以及最终收率高等优点。
技术实现要素:
[0013]
本发明的目的在于提供一种合成(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐的新方法。
[0014]
本发明的再一目的在于提供一种合成pf-07321332、波普瑞韦乃至paxlovid的新方法。
[0015]
在第一方面,本发明提供一种制备式1所示化合物的方法,所述方法如以下反应式所示:
[0016][0017]
所述方法包括以下步骤:
[0018]
1)二苯硫醚和2-溴丙烷发生反应得到式5所示化合物;
[0019]
2)式4所示化合物与式5所示化合物发生反应得到式6所示化合物;
[0020]
3)式6所示化合物在氯化氢作用下脱保护成盐,最终获得式1所示化合物。
[0021]
在具体的实施方式中,步骤1)在温度t1下,在溶剂s1中进行;
[0022]
所述溶剂s1为甲苯、二氯甲烷、四氢呋喃或它们的化合物;优选四氢呋喃;
[0023]
所述温度t1为-60℃~0℃;优选-25℃~-20℃。
[0024]
在具体的实施方式中,在步骤1)中,所述二苯硫醚与2-溴丙烷的摩尔比为1:1-1:5;优选1:1-1:3;更优选1:2。
[0025]
在具体的实施方式中,步骤2)在温度t2下,在溶剂s2中进行;
[0026]
所述溶剂s2为二氯甲烷、四氢呋喃、甲苯或它们的混合物;优选二氯甲烷;
[0027]
所述温度t2为-45℃~0℃;优选-25℃~-20℃。
[0028]
在具体的实施方式中,在步骤2)中,式4所示化合物与式5所示化合物的摩尔比为1:1-1:5;优选1:1-1:2;更优选1:1-1:1.3。
[0029]
在具体的实施方式中,步骤3)在温度t3下,在溶剂s3中进行;
[0030]
所述溶剂s3为乙酸乙酯、甲苯、甲基叔丁基醚或它们的混合物;优选甲基叔丁基醚;
[0031]
所述温度t3为20℃~65℃;优选40~65℃;更优选40℃~50℃。
[0032]
在具体的实施方式中,在步骤3)中,所述氯化氢是以气体形式通入反应体系。
[0033]
在优选的实施方式中,式4所示化合物与式5所示化合物反应生成式6所示化合物的收率在92%以上,优选94%以上,最优选96%以上。
[0034]
在优选的实施方式中,式6所示化合物在甲基叔丁基醚的氯化氢溶液脱保护得到最终式1所示化合物的收率在90%以上,优选93%以上,最优选97%以上。
[0035]
在优选的实施方式中,采用本发明方法得到式1所示化合物的总收率在82%以上,优选88%以上,最优选93%以上。
[0036]
在第二方面,本发明提供一种制备式7所示化合物的方法,
[0037][0038]
所述方法包括以下步骤:
[0039]
1)通过第一方面所述的方法制备式1所示化合物;和
[0040][0041]
2)利用步骤1)得到的式1所示化合物制备式7所示化合物。
[0042]
在第三方面,本发明提供一种制备paxlovid的方法,所述方法包括以下步骤:
[0043]
1)通过第一方面所述的方法制备式1所示化合物;
[0044][0045]
2)利用步骤1)得到的式1所示化合物制备式7所示化合物;和
[0046]
3)利用步骤2)得到的式7所示化合物制备paxlovid。
[0047]
在第四方面,本发明提供一种制备式8所示化合物的方法,
[0048][0049]
所述方法包括以下步骤:
[0050]
1)通过第一方面所述的方法制备式1所示化合物;和
[0051][0052]
2)利用式1所示化合物制备式8所示化合物。
[0053]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
[0054]
发明人经过广泛而深入的研究,出乎意料地发现在常规的(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐(式1所示化合物)的合成方法中,利用温和条件下合成的硫叶立德试剂替代膦叶立德试剂,不仅能够更安全地制备得到式1所示化合物,从而有利于其工业化生产,最终的式1所示化合物的收率也得到显著提高,从而为paxlovid和波普瑞韦的生产开辟了新的思路。在此基础上完成了本发明。
[0055]
本发明的方法
[0056]
为克服现有技术中合成(1r,2s,5s)-6,6-二甲基-3-氮杂双环[3,1,0]己基-2-羧酸甲酯盐酸盐(式1所示化合物)中存在的种种缺陷,本发明人以温和条件下合成的硫叶立德试剂替代膦叶立德试剂,从而提供了一种安全的、有利于工业化生产的式1化合物的合成方法。合成路线反应式如下所示:
[0057][0058]
在上述反应流程的基础上,本发明还对反应的工艺条件进行了优化,从而显著提高了反应收率。
[0059]
在具体的实施方式中,合成式5所示化合物时,溶剂s1为甲苯、二氯甲烷、四氢呋喃或它们的化合物;优选四氢呋喃;温度t1为-60℃~0℃;优选-25℃~-20℃;二苯硫醚与2-溴丙烷的摩尔比为1:1-1:5;优选1:1-1:3;更优选1:2。
[0060]
在具体的实施方式中,合成式6所示化合物时,溶剂s2为二氯甲烷、四氢呋喃、甲苯或它们的混合物;优选二氯甲烷;温度t2为-45℃~0℃;优选-25℃~-20℃;式4所示化合物与式5所示化合物的摩尔比为1:1-1:5;优选1:1-1:2;更优选1:1-1:1.3。
[0061]
在具体地实施方式中,合成式1所示化合物时,溶剂s3为乙酸乙酯、甲苯、甲基叔丁基醚或它们的混合物;优选甲基叔丁基醚;温度t3为20℃~65℃;优选40~65℃;更优选40℃~50℃;氯化氢是以气体形式通入反应体系。
[0062]
本领域技术人员熟知式1所示化合物是合成paxlovid的主要活性成分之一是为pf-07321332(式7化合物)以及波普瑞韦(式8化合物)的关键中间体,因此,基于本发明提供的合成式1所示化合物的方法,本发明人还提供了合成pf-07321332、波普瑞韦乃至paxlovid的新方法。
[0063]
基于本领域的现有技术文献,本领域技术人员可以知晓在利用本发明的方法合成得到式1所示化合物后,如何利进一步合成得到pf-07321332、波普瑞韦乃至paxlovid。
[0064]
本发明的优点:
[0065]
1.在本发明的方法中,式4所示化合物与硫叶立德试剂式5所示化合物反应生成式
6所示化合物的收率可以达到96%以上;
[0066]
2.在本发明的方法中,式6所示化合物在甲基叔丁基醚的氯化氢溶液脱保护得到最终式1所示化合物的收率为97%,总收率为93%以上。
[0067]
3.本发明的方法的工艺条件进行了优化,从而使得该方法不仅反应条件温和、有利于工业化生产,还提高了反应收率。
[0068]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0069]
实施例1:合成式4化合物
[0070]
3l的反应瓶中加入式2化合物367.5克(1.5摩尔),三氟甲磺酸酐465.5克(1.65摩尔),四氢呋喃1.5l,加完后室温反应2小时,tlc/hplc检测反应完全后,浓缩出去四氢呋喃,得到残余液加入2l的甲醇溶解,然后在0然下加入750g 20%的碳酸氢钾水溶液反应0.5小时后tlc/hplc检测反应完后蒸馏除去甲醇,残余的水层用甲苯600的甲苯萃取两次,合并甲苯层浓干得到式4化合物195.9克,收率86.3%。
[0071]
实施例2:合成式5化合物
[0072]
1l的反应瓶中加279.4克(1.5摩尔)的二苯硫醚,366克(3摩尔)的2-溴丙烷,加完后氮气保护下回流5小时。冷却蒸馏回收2-溴丙烷,残余液冷却室温后分为三等分到三个反应瓶中其中1号瓶子加入1l的二氯甲烷,2号瓶子加入1l的四氢呋喃,3号瓶子加入1l的甲苯,加完后三个反应瓶冷却到-25℃~-20℃,三个反应控温-25℃~-20℃分别加入168克(1.5摩尔)的叔丁醇钾,加完后三个反应分别反应30分钟,然后分别浓缩得式5化合物。计算三个反应的收率,如下表1。
[0073]
表1
[0074][0075]
实施例3:合成式5化合物
[0076]
1l的反应瓶中加279.4克(1.5摩尔)的二苯硫醚,366克(3摩尔)的2-溴丙烷,加完后氮气保护下回流5小时。冷却蒸馏回收2-溴丙烷,残余液冷却室温后分为三等分到三个反应瓶,三个反应瓶中分别加入1l的四氢呋喃中其中1号瓶子控温反应温度0℃~-5℃加入168克(1.5摩尔)的叔丁醇钾,2号瓶子控温反应温度-20℃~-25℃加入168克(1.5摩尔)的叔丁醇钾,3号瓶子控温反应温度-55℃~-60℃加入168克(1.5摩尔)的叔丁醇钾,,加完后三个反应分别反应30分钟,然后分别浓缩得式5化合物。计算三个反应的收率,如下表2。
[0077]
表2
[0078][0079]
实施例4:合成式5化合物
[0080]
取三个反应瓶子1号瓶子加入93.1克(0.5摩尔)的二苯硫醚,66.5克(0.5摩尔)的2-溴丙烷;2号瓶子加入93.1克(0.5摩尔)的二苯硫醚,133克(1摩尔)的2-溴丙烷;3号瓶子加入93.1克(0.5摩尔)的二苯硫醚,199.5克(1.5摩尔)的2-溴丙烷;三个反应加完后氮气保护下回流5小时。冷却蒸馏回收2-溴丙烷,残余液冷却室温后加入0.33升的四氢呋喃,加完后冷却到-25℃~-20℃,控温加入56克(0.5摩尔)的叔丁醇钾,加完后反应30分钟,然后浓缩得式5化合物。计算三个反应的收率,如下表3。
[0081]
表3
[0082][0083]
实施例5:合成式5化合物
[0084]
3l的反应瓶中加931.5克(5摩尔)的二苯硫醚,1220克(10摩尔)的2-溴丙烷,加完后氮气保护下回流5小时。冷却蒸馏回收2-溴丙烷,残余液冷却室温后加入3.3升的四氢呋喃,加完后冷却到-25℃~-20℃,控温加入560克(5摩尔)的叔丁醇钾,加完后反应30分钟,然后浓缩得式5化合物1190.7克收率97.7%。
[0085]
实施例6:合成式6化合物
[0086]
三个反应瓶中分别加入91.4克(0.4摩尔)式5化合物,然后1号反应瓶中加入333毫升的二氯甲烷,2号反应瓶中加入333毫升的甲苯,3号反应瓶中加入333毫升的四氢呋喃加完后三个反应品冷却到-20℃~-25℃,控温滴加入75.7克(0.33摩尔)的式4化合物,滴加完毕保温反应2小时,tlc/hplc检测反应完后,反应液加入1n的稀硫酸,搅拌分层,水层用二氯甲烷萃取一次合并有机层干燥浓缩得到式6化合物。计算三个反应的收率,如下表4。
[0087]
表4
[0088][0089]
[0090]
实施例7:合成式6化合物(优化反应温度)
[0091]
三个反应瓶中分别加入91.4克(0.4摩尔)式5化合物和333毫升的四氢呋喃加完后1号反应瓶降温到0℃~-5℃,2号反应瓶降温到-20℃~-25℃,3号反应瓶降温到-40℃~-45℃;然后1号反应控制温度0℃~-5℃滴加入75.7克(0.33摩尔)的式4化合物;2号反应控制温度-20℃~-25℃滴加入75.7克(0.33摩尔)的式4化合物;3号反应控制温度-40℃~-45℃滴加入75.7克(0.33摩尔)的式4化合物;滴加完毕三个反应保温反应2小时,tlc/hplc检测反应完后,反应液加入1n的稀硫酸,搅拌分层,水层用二氯甲烷萃取一次合并有机层干燥浓缩得到式6化合物。计算三个反应的收率,如下表5。
[0092]
表5
[0093][0094]
实施例8:合成式6化合物
[0095]
取三个反应瓶,1号反应品加入75.44克(0.33摩尔)式5化合物和333毫升的四氢呋喃;2号反应品加入94.44克(0.4摩尔)式5化合物和333毫升的四氢呋喃;3号反应品加入114.3克(0.5摩尔)式5化合物和333毫升的四氢呋喃;加完后三个反应降温到-25℃~-20℃,然后控温滴加入75.7克(0.33摩尔)的式4化合物,滴加完毕保温反应2小时,tlc/hplc检测反应完后,反应液加入1n的稀硫酸,搅拌分层,水层用二氯甲烷萃取一次合并有机层干燥浓缩得到式6化合物。计算三个反应的收率,如下表6。
[0096]
表6
[0097][0098][0099]
实施例9:合成式6化合物
[0100]
2l的反应瓶中加入274.1克(1.2摩尔)式5化合物,1升的二氯甲烷,加完后冷却到-25℃~-20℃,控温滴加入227.3克(1摩尔)的式4化合物,滴加完毕保温反应2小时,tlc/hplc检测反应完后,反应液加入1n的稀硫酸,搅拌分层,水层用二氯甲烷萃取一次合并有机层干燥浓缩得到式6化合物260.7克,收率96.8%。
[0101]
实施例10:合成式1化合物
[0102]
取三个反应瓶,分别加入170克(0.65摩尔)的式6化合物,加完后1号反应瓶加入乙酸乙酯750毫升;2号反应瓶加入甲苯750毫升;3号反应瓶加入甲基叔丁基醚750毫升;然后三个反应瓶分别计量通入26.5克(0.715摩尔)的氯化氢气体通完后40-45℃反应3小时,过滤,滤饼真空干燥得到类白色的式1化合物。计算三个反应的收率,如下表7。
[0103]
表7
[0104][0105]
实施例11:合成式1化合物
[0106]
取三个反应瓶,分别加入170克(0.65摩尔)的式6化合物和甲基叔丁基醚750毫升,然后分别向三个反应器中通入26.5克(0.715摩尔)的氯化氢气体。通完后1号反应器20-25℃反应3小时;2号反应器40-45℃反应3小时;3号反应器60-65℃反应3小时;最后三个反应分别过滤,滤饼真空干燥得到类白色的式1化合物。计算三个反应的收率,如下表8。
[0107]
表8
[0108][0109][0110]
实施例12:合成式1化合物
[0111]
340克(1.3摩尔)的式6化合物加入1.5升的甲基叔丁基醚溶解,然后计量通入53克(1.43摩尔)的氯化氢气体通完后40-45℃反应3小时,过滤,滤饼真空干燥得到类白色的式1化合物198.6克。收率96.9%。
[0112]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1