硝酮嗪晶型、制备方法及应用与流程
本发明涉及医药化学领域,具体涉及硝酮嗪晶型、制备方法及其应用。
背景技术:
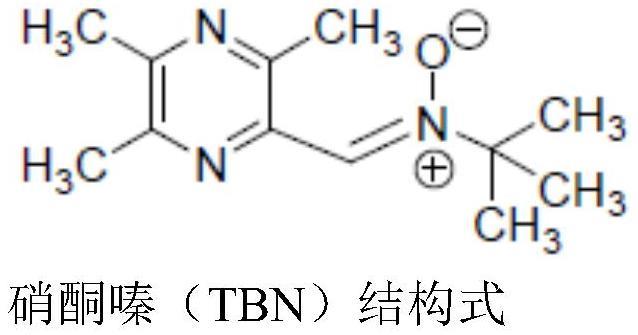
1、硝酮嗪是川芎嗪(tmp)的硝酮衍生物,是在川芎嗪的结构基础上增加硝酮药效基团经化学合成的全新化合物,简称为tbn,它的化学名为(顺)-2-甲基-n-[(3,5,6-三甲基吡嗪-2-)次甲基]2-丙胺氧化物,分子式为c12h19n3o,分子量是221.30,具有下式所示的化学结构:
2、
3、硝酮嗪可以抑制缺血所致的神经细胞氧化损伤,从而发挥神经细胞保护作用,缓解脑栓塞伴随的神经症状及功能障碍。临床上可用于神经系统疾病、心脑血管疾病及退行性老化疾病等的治疗。
技术实现思路
1、本发明目的在于提供硝酮嗪晶型、制备方法及其应用。
2、本发明的目的之一在于提供一种硝酮嗪的晶型a,所述的晶型其xrpd图谱在2θ角度位置为:10.60±0.2、11.03±0.2、15.31±0.2、15.55±0.2、17.14±0.2、17.93±0.2、23.81±0.2处有特征衍射峰。
3、在一种具体的实施方案中,硝酮嗪的晶型a,所述的晶型其xrpd图谱在2θ角度位置为:10.60±0.2、11.03±0.2、13.51±0.2、15.31±0.2、15.55±0.2、17.14±0.2、17.93±0.2、21.22±0.2、23.81±0.2、25.23±0.2、27.08±0.2处有特征衍射峰。
4、在一种具体的实施例中,硝酮嗪的晶型a具有与说明书图1基本上相同的xrpd图谱。
5、进一步的,本发明所述的硝酮嗪晶型a有针状、方块状、棒状三种晶态,显微照片如图5和图6。
6、进一步的,本发明所述的硝酮嗪晶型a熔点分别在76℃-78℃。
7、进一步的,本发明所述的硝酮嗪晶型a的dsc图谱基本如图3。
8、进一步的,本发明所述的硝酮嗪晶型a的tga图谱如图7。
9、进一步的,本发明提供了硝酮嗪晶型a红外图谱,如图9。
10、本发明经过系统的晶型筛选实验,其中筛选方法包括蒸发结晶法(单一溶剂法和混合溶剂法),热溶冷析法和混悬打浆法等。筛选包括四氢呋喃、乙酸乙酯、甲苯、丙酮、二氧六环、异丙醇、石油醚、正己烷、乙酸异丙酯、异辛烷和乙酸异丁酯等溶剂,筛选结果均为晶型a。
11、进一步的,本发明还提供了一种硝酮嗪晶型a的制备方法,包括以下步骤:
12、(1)将硝酮嗪粗品与有机溶剂混合,水浴加热至60~80℃,搅拌过滤,滤液冷却结晶得晶体状固体;
13、(2)步骤(1)得到的晶体状固体和正庚烷混合加热溶解,冷却结晶得硝酮嗪晶型a。
14、进一步的,本发明所述的制备方法,步骤(1)中所述的有机溶剂选自乙酸乙酯、正己烷、正庚烷及环己烷中的一种或几种,进一步优选的,有机溶剂为正己烷或正庚烷;或者所述有机溶剂选自正己烷与乙酸乙酯的混合溶剂,申请人发现,选用优选的有机溶剂可以显著降低杂质含量。
15、进一步的,本发明所述的制备方法,步骤(1)中硝酮嗪粗品与有机溶剂的重量体积比为1:5~20,优选1:8~12。
16、进一步的,本发明所述的制备方法,步骤(1)中冷却结晶的温度选自2~12℃,更优选3~10℃,最优选为3~5℃,申请人发现,在最优选的范围内,可以显著降低杂质含量。
17、进一步的,本发明所述的制备方法,步骤(2)中硝酮嗪粗品与正庚烷的重量体积比优选1:1~5,最优选1:1~3。
18、进一步的,本发明所述的制备方法,步骤(2)中晶体状固体和正庚烷混合加热温度为60~80℃,优选65~75℃。
19、进一步的,本发明所述的制备方法,步骤(2)中冷却结晶的温度选自2~12℃,更优选4~10℃。
20、本发明的目的之二在于提供一种硝酮嗪的二水合物,其xrpd图谱在2θ角度位置为:8.91±0.2、11.46±0.2、14.29±0.2、17.60±0.2、21.19±0.2、22.02±0.2、23.19±0.2、24.30±0.2、24.92±0.2、29.20±0.2、31.41±0.2处有特征衍射峰。
21、在一种具体的实施方案中,硝酮嗪的二水合物,其xrpd图谱在2θ角度位置为:8.91±0.2、11.46±0.2、12.00±0.2、14.29±0.2、17.60±0.2、19.50±0.2、21.19±0.2、22.02±0.2、23.19±0.2、24.30±0.2、24.92±0.2、26.70±0.2、29.20±0.2、31.41±0.2、36.20±0.2处有特征衍射峰。
22、在一种具体的实施例中,硝酮嗪的二水合物具有与说明书图2基本上相同的xrpd图谱。
23、进一步的,本发明所述的硝酮嗪的二水合物熔点在37–40℃。
24、进一步的,本发明所述的硝酮嗪的二水合物的dsc图谱基本如图4。
25、进一步的,本发明所述的硝酮嗪的二水合物的tga图谱如图8。
26、本发明所述的硝酮嗪二水合物在达到熔点时(约37–40℃)重量开始发生减少,失重13.67%。
27、本发明所述的硝酮嗪的二水合物是硝酮嗪在饱和的乙醇水溶液二元体系中冷析得到,优选乙醇水溶液中乙醇体积百分比为5~50%,更优选为5~20%。
28、在另一方面,本发明提供一种药物组合物,其包含一种或多种本发明的晶型a或二水合物。所述药物组合物还可以任选地包含药学上可接受的载体、赋形剂、填充剂、粘合剂、崩解剂、助流剂和/或介质等。
29、在另一方面,本发明所述的晶型a和/或二水合物,或者本发明所述的药物组合物可以通过口服或注射等给药途径。
30、在另一方面,本发明还提供一种包含本发明的晶型a和/或二水合物的剂型,包括但不限于片剂、胶囊剂、粉针剂、分散剂等,优选片剂和粉针剂。
31、在另一方面,本发明还提供本发明所述的晶型a和/或二水合物或药物组合物在用于制备治疗神经系统疾病、心脑血管疾病及退行性老化疾病药物中的应用。
32、本发明的有益效果:
33、(1)本发明所述的硝酮嗪晶型a和硝酮嗪二水合物制备方法简单,易于工业化大生产。
34、(2)本发明所述的硝酮嗪晶型a在热处理、机械处理和加速(50℃,75%rh)稳定的为晶型a,转晶风险小,稳定性好,有利于制剂在制备及其储存过程中的稳定性,有效保证制剂中晶型含量的一致性,安全、有效、质量可控。
35、(3)本发明所述的硝酮嗪晶型a在大部分溶剂中(如乙腈、甲醇、乙醇、丙酮和乙酸乙酯等)易溶,在水中极易溶解,有较高的生物活性和良好的成药性,生物利用度高,药效发挥快,具有较高的生物活性。
技术特征:
1.一种硝酮嗪的晶型a,其特征在于,所述的晶型其xrpd图谱在2θ角度位置为:10.60±0.2、11.03±0.2、15.31±0.2、15.55±0.2、17.14±0.2、17.93±0.2、23.81±0.2处有特征衍射峰。
2.如权利要求1所述硝酮嗪的晶型a,其特征在于,所述的晶型a其xrpd图谱在2θ角度位置为:10.60±0.2、11.03±0.2、13.51±0.2、15.31±0.2、15.55±0.2、17.14±0.2、17.93±0.2、21.22±0.2、23.81±0.2、25.23±0.2、27.08±0.2处有特征衍射峰。
3.根据权利要求1所述的晶型a,其特征在于,具有与图1基本上相同的xrpd图谱。
4.根据权利要求1所述的晶型a,其特征在于,a熔点在76℃-78℃。
5.根据权利要求1所述的晶型a,其特征在于,dsc图谱基本如图3,tga图谱基本如图7,红外图谱基本如图9。
6.权利要求1~5任一项所述晶型a的制备方法,其特征在于,包括以下步骤:
7.根据权利要求6所述的制备方法,其特征在于,步骤(1)中所述的有机溶剂选自乙酸乙酯、正己烷、正庚烷及环己烷中的一种或几种。
8.根据权利要求6或7所述的制备方法,其特征在于,步骤(1)中硝酮嗪粗品与有机溶剂的重量体积比为1:5~20,优选1:8~12;步骤(1)中冷却结晶的温度选自2~12℃,更优选3~10℃,最优选为3~5℃;步骤(2)中硝酮嗪粗品与正庚烷的重量体积比优选1:1~5,最优选1:1~3;步骤(2)晶体状固体和正庚烷混合加热温度为60~80℃,优选65~75℃;冷却结晶的温度选自2~12℃,更优选4~10℃。
9.一种药物组合物,其特征在于,包含权利要求1~5任一项所述的晶型a。
10.根据权利要求1~5任一项所述的晶型a在制备治疗神经系统疾病、心脑血管疾病及退行性老化疾病药物中的应用。
技术总结
本发明公开硝酮嗪晶型、制备方法及其应用,其中本发明提供一种硝酮嗪的晶型A,其XRPD图谱在2θ角度位置为:10.60±0.2、11.03±0.2、15.31±0.2、15.55±0.2、17.14±0.2、17.93±0.2、23.81±0.2处有特征衍射峰。所述的晶型A转晶风险小,稳定性好,有利于制剂在制备及其储存过程中的稳定性,有效保证制剂中晶型含量的一致性,安全、有效、质量可控,有较高的生物活性和良好的成药性,生物利用度高,药效发挥快,具有较高的生物活性。
技术研发人员:刘伟,孙业伟,王玉强
受保护的技术使用者:广州喜鹊医药有限公司
技术研发日:
技术公布日:2024/1/11
- 还没有人留言评论。精彩留言会获得点赞!