一种用于红球菌基因敲除的重组系统及其应用
1.本发明属于细胞工程技术领域,具体涉及一种用于红球菌基因敲除的重组系统及其应用。
背景技术:
2.红球菌属(rhodococcus)是一种需氧的、非运动的、无芽孢的和细胞壁含有分枝酸的革兰氏阳性细菌,介于分枝杆菌属(mycobacterium)、诺卡氏菌属(nocardia)和棒状杆菌属(corynebacterium)之间。红球菌广泛分布于从海洋到陆地的不同环境中,能适应恶劣环境,具有多种有机物的降解代谢活性,如单芳烃、多芳烃、酚类、卤代烃、硝基芳香烃、腈和其它芳香族化合物。虽然人们开始认识到红球菌在生物降解、转化和合成等领域的应用潜力,但是红球菌基础研究的数量却远不及大肠杆菌和芽孢杆菌等较为成熟的工程菌。人们对红球菌的了解并未深入。而红球菌的基因敲除工具对于人们开展红球菌的基础研究非常重要。
3.目前,红球菌可用的基因敲除工具非常少,而且现有的传统同源重组技术在红球菌的基因敲除中表现出重组效率低的特点。现有研究发现通过分枝杆菌噬菌体重组酶che9c60&61可以准确、高效地对红球菌基因进行编辑,但是这种方式会在基因敲除的过程中引入抗生素抗性基因,导致无法进行多轮基因编辑。
4.因此,研发一种重组效率高,且可以进行多轮基因编辑的可以用于红球菌基因敲除的重组系统是有必要的。
技术实现要素:
5.为了解决上述问题,本发明目的之一在于提供一种用于红球菌基因敲除的重组系统。该重组系统基于frt位点特异性重组酶flpw方法可有效对红球菌进行高效无痕敲除,为下一轮基因编辑(多基因敲除)回收了抗性基因。
6.为了达到上述目的,本发明可以采用以下技术方案:
7.本发明一方面提供一种用于红球菌基因敲除的重组系统,其包括:che9c60&61重组酶质粒、flp重组酶和游离双链dna敲除盒片段;游离双链dna敲除盒片段包括:目的基因上下游同源臂序列及在其间插入包含抗性基因的两个同向frt序列;flp重组酶质粒的基因序列如seq id no.1或seq id no.3所示。
8.本发明另一方面提供一种基因工程红球菌的构建方法,包括:使用上述用于红球菌基因敲除的重组系统对红球菌进行基因敲除得基因工程红球菌。
9.本发明再一方面提供一种上述的基因工程红球菌的构建方法构建的基因工程红球菌。
10.本发明再一方面提供一种flp重组酶基因序列,其包括如seq id no.1或seq id no.3所示序列。
11.本发明再一方面提供一种上述用于红球菌基因敲除的重组系统或上述的flp重组
酶基因序列在红球菌基因编辑中的应用。
12.本发明有益效果包括:本发明提供的用于红球菌基因敲除的重组系统可以有效对红球菌进行无痕敲除,庆大霉素抗性丢失效率达到了100%和78.7%;而且本发明基于frt位点特异性重组酶flpw方法可有效实现红球菌rhodococcus ruber r1的高效无痕敲除,为下一轮基因编辑(多基因敲除)回收抗性基因。
附图说明
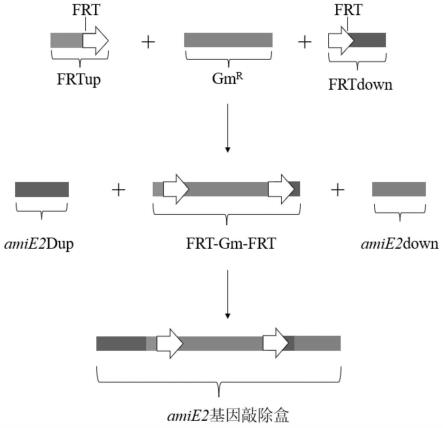
13.图1为amie2基因敲除盒片段构建示意图;
14.图2为pbnvw质粒构建示意图;
15.图3为rhodococcus ruber r1与其突变株pcr验证;
16.图4为rhodococcusruber r1与其突变株测序验证;
17.图5为rhodococcus ruber r1δamie2::frt-gm-frt的抗性丢失点板筛选;
18.图3中,注:泳道1、2、3是依次以rhodococcus ruber r1、rhodococcus ruber r1δamie2::frt-gm-frt、rhodococcus ruber r1δamie2为模板,amie2-p9/p10引物扩增出的dna片段;m表示marker。
具体实施方式
19.所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
20.本文中使用的术语仅用于描述特定实施例,并且无意于限制本公开。除非在上下文中具有明显不同的含义,否则单数形式的表达包括复数形式的表达。如本文所使用的,应当理解,诸如“包括”、“具有”、“包含”之类的术语旨在指示特征、数字、操作、组件、零件、元件、材料或组合的存在。在说明书中公开了本发明的术语,并且不旨在排除可能存在或可以添加一个或多个其他特征、数字、操作、组件、部件、元件、材料或其组合的可能性。如在此使用的,根据情况,“/”可以被解释为“和”或“或”。
21.本发明一实施例提供一种用于红球菌基因敲除的重组系统,其包括:che9c60&61重组酶质粒、flp重组酶和游离双链dna敲除盒片段;游离双链dna敲除盒片段包括:目的基因上下游同源臂序列及在其间插入包含抗性基因的两个同向frt序列;flp重组酶质粒的基因序列如seq id no.1或seq id no.3所示。
22.需要说明的是,seq id no.1所示序列是flp基因序列,seq id no.3所示序列进行红球菌密码子优化(也称flpw基因),并在flpw基因起始密码子前添加组成型pamic启动子序列;在pamic的5’端设计xbaⅰ酶切位点,在flpw的3’端设计kpnⅰ酶切位点,得到5’xba
ⅰ‑
pamic-flpw-kpnⅰ3’序列(即seq id no.3)。
23.还需要说明的是,本发明发现在抗生素抗性基因的两端引入同向frt位点,再利用位点特异性重组酶flp对两个同向frt位点进行准确、高效重组,使frt位点之间的抗性基因丢失,就能达到无痕敲除并且可以进行多轮基因编辑的目的。
24.还需要说明的是,本发明实施例以puc57为载体,构建携带包含上述敲除盒片段的质粒,再利用pcr扩增或者限制性内切酶消化该质粒,得到所需要的游离双链dna敲除盒片
段。该片段将用于无痕基因敲除的重组系统使用,并在rhodococcus ruber r1中验证了该系统的可行性。
25.在一些具体实施例中,上述用于红球菌基因敲除的重组系统中,抗性基因包括庆大霉素抗性基因。需要说明的是,抗性基因可以为本领域所已知的,比如庆大霉素抗性基因。
26.本发明另一实施例提供一种基因工程红球菌的构建方法,包括:使用上述用于红球菌基因敲除的重组系统对红球菌进行基因敲除得基因工程红球菌。红球菌优选为rhodococcus ruber r1;红球菌基因优选为δamie2。
27.在一些具体实施例中,上述基因工程红球菌的构建方法中,包括:在宿主中过表达che9c60&61重组酶基因,并利用同源重组的方法把两个同向frt间包含抗性基因的序列重组到红球菌的目的敲除基因上;再利用优化flp重组酶实现抗性基因的丢失。
28.本发明再一实施例提供一种上述的基因工程红球菌的构建方法构建的基因工程红球菌。
29.本发明再一实施例提供一种flp重组酶基因序列,其包括如seq id no.1或seq id no.3所示序列。需要说明的是,本发明中的flp重组酶基因序列编码的重组酶可介导红球菌基因组中目的基因敲除位置的两个同向frt位点的高效重组。
30.本发明再一实施例提供一种上述用于红球菌基因敲除的重组系统或上述的flp重组酶基因序列在红球菌基因编辑中的应用。需要说明的是,可以将上述的用于红球菌基因敲除的重组系统或上述的flp重组酶基因序列用于红球菌基因的敲除,重组效率高,且可以进行多轮基因编辑。
31.为了更好地理解本发明,下面结合具体示例进一步阐明本发明的内容,但本发明的内容不仅仅局限于下面的示例。
32.本发明实施例中,使用的红球菌rhodococcus ruber r1、大肠杆菌dh5α为本单位保藏;使用的质粒prctc-pa2-che9c60&61、pbnvcm、puc57、pkd3、psrkgm和psrkkm为本单位保藏;限制性内切酶、连接酶等试剂购自宝日医生物技术(北京)有限公司;一步快速克隆和taq酶购自北京金沙生物科技有限公司。
33.本发明实施例中,pcr引物由广州艾基生物技术有限公司合成;质粒pbnvw和puc57w1为本发明构建。
34.本发明实施例中,选择酰胺酶基因amie2作为敲除目的基因,该基因敲除后可提高菌株丙烯酰胺的产量。
35.本发明实施例中,使用的的引物对如下表1所示(划线部分为酶切位点)。
36.表1引物序列
[0037][0038]
本发明实施例中,rhodococcus ruber r1δamie2::frt-gm-frt(seq id no.4)和frt重组后的rhodococcus ruber r1δamie2序列(seq id no.5),其中,下划线为amie2-p9/p10引物序列,大写字母为amie2上下游基因序列,小写字母为敲除盒内非同源臂序列,字符底纹加框区域为frt序列:
[0039]
seq id no.4(rhodococcus ruber r1δamie2::frt-gm-frt):
[0040][0041][0042]
seq id no.5(rhodococcus ruber r1 δamie2):
[0043][0044]
(1)rhodococcus ruber r1/prctc-pa2-che9c60&61构建
[0045]
为使prctc-pa2-che9c60&61(liang et al.metabolic engineering,2020,57:13-22)导入rhodococcus ruber r1中,首先制备rhodococcus ruber r1的电击转化感受态细胞。将rhodococcus ruber r1混合在10ml电击准备液(20g葡萄糖、5g酵母粉、0.655g k2hpo4·
3h2o、0.5g kh2po4、0.5g mgso4·
7h2o、8.5g甘氨酸和0.0015g异烟肼,使用双蒸水定容至1l)中;
[0046]
另外,电击转化双链dna敲除盒片段时需要在上述10ml电击准备液中添加5mg尿素,在30℃和200rpm培养2d-3d,取1ml菌液至冰上预冷5min,4℃6000rpm离心5min,去上清液,沉淀用4℃预冷的10%(v/v)甘油1ml重悬,该步骤重复3次;最后一次离心去上清后,沉淀用100μl的10%甘油进行重悬,加入1μg左右的游离双链dna敲除盒片段,在冰上静置5min;把100μl混合质粒的菌液加入1mm电击杯中,设定电转化仪电压1250v进行电击转化;取出电击杯中100μl的菌液至1.5ml离心管中,加入900μl电击复苏液(5蛋白胨、5g氯化钠、2.5g酵母提取物、18.5g脑心浸液和91g山梨糖醇,双蒸水定容至1l),在30℃、200rpm培养3h-4h,涂布至四环素(10μg/ml)lb平板30℃培养3d-4d。
[0047]
菌落pcr验证后得rhodococcus ruber r1/prctc-pa2-che9c60&61,并用上述制备电击感受态的方法的制备感受态细胞,用于双链dna敲除盒片段的电击转化。
[0048]
(2)puc57-amie2up-down质粒构建
[0049]
首先以puc57为载体,构建包含amie2基因敲除盒的质粒:puc57w1(敲除盒构建过程如图1所示)。使用菌株rhodococcus ruber r1的amie2基因设计引物:amie2-p1/p2/p3/p4/p5/p6/p7/p8(见表1);分别用amie2-p5/p2和amie2-p6/p3引物片段扩增获得amie2基因的上下游约700bp片段(50μl扩增体系:amie2-p5/p2或amie2-p6/p3引物片段各2μl,rhodococcus ruber r1总dna 1μl,s4 fidelity pcr mix 45μl;扩增条件:98℃预变性2min,98℃变性10s,58℃退火15s,72℃延伸15s;程序循环数:35个循环)。
[0050]
本发明实施例在设计引物时,预先在amie2-p2/p3引物末端设计了包含有speⅰ和bamhⅰ两个酶切位点的短重叠延伸连接头,利用重叠延伸pcr方法,先用上述扩增获得的两段约700bp和900bp的片段互为引物,重叠延伸pcr扩增10个循环(25μl扩增体系:上述两段约700bp的片段各2μl,s4 fidelity pcr mix 19μl;扩增条件:98℃预变性2min,98℃变性10s,54℃退火15s,72℃延伸15s;程序循环数:10个循环);
[0051]
再以重叠延伸pcr产物为模板,用amie2-p1/p4为引物扩增可将amie2基因的上游基因和下游基因各约500bp拼接起来(50μl扩增体系:amie2-p1/p4引物片段各2μl,重叠延伸pcr产物1μl,s4 fidelity pcr mix 45μl;扩增条件:98℃预变性2min,98℃变性10s,58℃退火15s,72℃延伸25s;程序循环数:35个循环);
[0052]
将重叠延伸后片段用限制性内切酶hindⅲ和ecorⅰ消化后(酶切条件为:片段和质粒50μl,10
×
buffer 10μl,hindⅲ2μl,ecorⅰ2μl,补足双蒸水至100μl,37℃反应2h),连接至以同样内切酶消化过的puc57(连接体系为:10
×
连接酶buffer 1μl,酶切后片段6μl,酶切后载体2μl,t4 dna连接酶1μl双蒸水补至20μl,16℃连接12h),热激转化dh5α并菌落pcr验证后,获得puc57-amie2up-down质粒。
[0053]
(3)构建携带两个同向frt间包含庆大霉素抗性基因的序列的质粒
[0054]
以pkd3中的frt序列为模板,frt-p1/p2/p3/p4为引物pcr扩增获得两个片段(frtup和frtdown),用一步快速克隆法构建psrkkm-frtup-down质粒(一步快速克隆体系为:psrkkm 1μl,frtup和frtdown各2μl,2
×
uniclone seamless cloning mix 5μl;反应条件为:50℃处理20min,冰浴2min);热激转化dh5α并菌落pcr验证后,获得psrkkm-frtup-down质粒。
[0055]
以psrkgm质粒中的庆大霉素抗性基因序列为模板,gm-p1/p2为引物pcr扩增获得庆大霉素抗性基因片段,使用一步快速克隆法把上述得到的庆大霉素抗性基因片段重组到psrkkm-frtup-down质粒上(一步快速克隆体系为:psrkkm-frtup-down 1.5μl,庆大霉素抗性基因片段3.5μl,2
×
uniclone seamless cloning mix 5μl;反应条件为:50℃处理20min,冰浴2min),热激转化dh5α并菌落pcr验证后,获得psrkkm-frt-gm-frt质粒。
[0056]
(4)rhodococcus ruber r1δamie2::frt-gm-frt/prctc-pa2-che9c60&61突变株构建
[0057]
以psrkkm-frt-gm-frt和puc57-amie2up-down设计用于一步快速克隆引物amie2-frt-gm-p1/p2,用pcr扩增获得1142bp的片段,根据上述方法使用一步快速克隆获得frt-gm-frt片段插入在puc57-amie2up-down中间的puc57w1质粒。通过amie2-p7/p8引物pcr扩增或者用限制性内切酶hindⅲ和ecorⅰ消化puc57w1可以得到amie2基因的游离双链dna敲除盒片段。
[0058]
把amie2的双链dna敲除盒片段通过电击转化到rhodococcus ruber r1/prctc-pa2-che9c60&61后,用amie2-p9/p10引物菌落pcr和测序验证后(结果图3和图4所示),得到rhodococcus ruber r1δamie2::frt-gm-frt/prctc-pa2-che9c60&61突变株(见seq id no.4)。
[0059]
(5)pbnvw质粒构建
[0060]
为得到一个有高效质粒退出机制并携带flpw重组酶的质粒,需要以携带在可在37℃高效退出的温敏复制子pb264的pbnvcm质粒为载体,连接优化重组酶flpw(重组质粒名为pbnvw)。委托南京金斯瑞生物科技有限公司对flp基因(见seq id no.1)编码的重组酶flp的氨基酸序列(见seq id no.2)进行红球菌密码子优化(优化所得到基因命名为flpw基因),并在flpw基因起始密码子前添加组成型pamic启动子序列。在pamic的5’端设计xbaⅰ酶切位点,在flpw的3’端设计kpnⅰ酶切位点,由南京金斯瑞生物科技有限公司人工合成5’xba
ⅰ‑
pamic-flpw-kpnⅰ3’序列(见seq id no.3),并通过同样的酶切位点重组到pbnvcm载体上,获得pbnvw质粒(质粒构建示流程如图2所示)。
[0061]
为达到无痕敲除的目的,需要使用flpw重组酶对rhodococcus ruber r1δamie2::frt-gm-frt的两个同向frt位点发生重组,使庆大霉素抗性丢失,为此需要制备rhodococcus ruber r1δamie2::frt-gm-frt的电击感受态细胞。
[0062]
使用上述制备电击感受态的方法制备rhodococcus ruber r1δamie2::frt-gm-frt/prctc-pa2-che9c60&61的电击感受态细胞。电击转化完成后,用pb264-p1/p2引物进行菌落pcr验证。把已验证的rhodococcus ruber r1δamie2::frt-gm-frt/prctc-pa2-che9c60&61/pbnvw在5ml液体lb培养基中30℃,200rpm培养2-3d后,取100μl菌液,稀释至10-6
涂布在四环素和氯霉素(15μg/ml)lb平板上培养3-4d。用无菌牙签挑取单菌落依次点到庆大霉素lb平板和氯霉素lb平板上,由于flpw重组酶对frt位点发生作用使庆大霉素抗性丢失的效率不同,所以会产生frt位点没有发生重组和frt位点发生重组的两种菌株。不能在庆大霉素lb平板上生长的菌株为frt位点发生重组使庆大霉素抗性丢失的rhodococcus ruber r1δamie2突变株(见seq id no.5)。
[0063]
(6)flpw效率验证
[0064]
用amie2-p9/p10引物进行菌落pcr和测序验证(结果如图3和图4所示),得到rhodococcus ruber r1δamie2/prctc-pa2-che9c60&61/pbnvw。为验证flpw的效率,本发明实施例对rhodococcus ruber r1δamie2::frt-gm-frt菌株进行了两次pbnvw的电击转化,每次电击转化挑取3个pbnvw阳性转化子根据上述实验步骤进行抗性负筛选并进行数据记录。其中庆大霉素抗性丢失效率是flpw对rhodococcus ruber r1δamie2::frt-gm-frt中frt位点重组效率的表现,代表优化重组酶flpw在rhodococcus ruber r1中的效率(统计数据结果如图5和表2所示)。从图5和表1中可以看出两次电击pbnvw介导rhodococcus ruber r1δamie2::frt-gm-frt菌株的无痕敲除,庆大霉素抗性丢失效率达到了100%和78.7%,突变株的序列分析表明flpw介导两个frt位点之间的重组,从而删除frt位点之间庆大霉素抗性基因,只留下一个frt位点(见seq id no.5)。这说明本发明建立的基于frt位点特异性重组酶flpw方法可实现红球菌rhodococcus ruber r1的高效无痕敲除,为下一轮基因编辑(多基因敲除)回收了抗性基因。
[0065]
表2优化重组酶flpw在rhodococcus ruber r1中的效率
[0066][0067][0068]
把已验证的rhodococcusruberr1δamie2/prctc-pa2-che9c60&61/pbnvw在37℃培养并丢失pbnvw质粒,用pb264-p1/p2引物进行菌落pcr验证。得到rhodococcusruberr1δamie2/prctc-pa2-che9c60&61可用于下一轮基因编辑(多基因敲除)。完成最后一轮基因编辑后,可将prctc-pa2-che9c60&61质粒通过连续传代丢失,得到最终的基因工程菌,方法如下:将编辑成功的红球菌接种于不含抗生素的5mllb培养基中,于30℃、200rpm培养2d-3d,取菌液适当稀释后涂布于不含抗生素的平板,30℃培养2d-3d,生长的菌落分别划线于无抗性lb平板和四环素(10μg/ml)lb平板,若在四环素lb平板上不生长说明prctc-pa2-che9c60&61质粒已丢失,挑取相应菌落进行培养,得到的红球菌工程菌株。
[0069]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1