一种阿维巴坦中间体的制备方法
本发明属于药物化学合成,具体涉及一种抗生素药物阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸乙酯草酸盐的制备方法。
背景技术:
1、阿维巴坦(avibactam,nxl-104)属于二氮杂双环辛酮化合物,是目前最被看好的新型β-内酰胺酶抑制剂。与三种已上市的β-内酰胺酶抑制剂相比,具有长效和与酶可逆性共价结合,且不会诱导β-内酰胺酶产生。2015年2月25日美国fda批准艾尔健公司的新型抗生素复方药物阿维巴坦头孢他啶(avibactam-ceftazidime),商品名为avycaz。阿维巴坦-头孢他啶联合甲硝唑用于治疗复杂性腹腔内感染(ciai)及复杂性尿路感染(cuti)。
2、阿维巴坦钠(avibactam sodium)又被称为nxl-104,属于二氮杂双环辛烷类化合物。化学名为硫酸单[(1r,2s,5r)-2-氨基羰基-7-氧代-1,6-氮杂双环[3.2.1]辛-6-基]酯钠盐,具体的化学结构如结构式所示,该化合物以五并六元环系为骨架,同时含有2s和5r两个手性中心,五元环中的酰胺键是β-内酰胺酶的作用位点,可以打开和闭合,因此能够起到长时间的抑酶效果。
3、
4、目前,阿维巴坦钠合成路线大致可分为:
5、(1)以l-焦谷氨酸衍生物为起始原料合成阿维巴坦钠。如scheme 1所示,专利wo2012086241a1报道了以cbz保护的l-焦谷氨酸1为起始原料,经过亲电加成、硫叶立德开环增碳、催化合环、硼氢化钠还原、脱cbz保护得到关键中间体6,再通过酰化保护氨基、苄氧胺取代、脱保护、分子内合脲环、水解、氨解、脱苄基、磺化、季铵盐络合、成钠盐共15步反应得到阿维巴坦钠,反应总收率为9.8%。该路线反应步骤多、反应所需时间较长,且每步中间体都需要柱层析分离纯化,操作繁琐,总收率也低,故不适合工业化生产。
6、
7、wo2009090320a1开发了另外一条以l-焦谷氨酸衍生物为起始原料的合成路线,如scheme 2所示。该路线以双保护的l-焦谷氨酸为原料,首先利用硫叶立德开环增碳,再用氯化锂氯代,苄氧胺盐酸盐上苄氧胺得17,之后甲磺酸脱boc保护,碳酸氢钾合环,三乙酰基硼氢化钠还原碳氮双键,草酸分离得关键中间体19,三光气合脲环构成五并六元环系,氢氧化锂水解酯键得到想要的5r构型化合物12,最后通过氢化脱苄基保护、磺化上磺酸基团,再与季铵盐络合,与异辛酸钠交换得到终产物阿维巴坦钠14。
8、
9、中国专利cn1468242报道了以手性哌啶环衍生物21为原料,经过三氟乙酸酐的酰化保护、与苄氧胺的反式亲核取代、硼氢化钠还原脱保护、三光气合脲环、四三苯基膦钯催化除去烯丙基、氨取代得到关键中间体12,再pd/c氢化、三氧化硫吡啶磺化、四丁基铵盐络合、最后成钠盐得到终产物14,如scheme 3所示。
10、
11、综上所述,(2s,5r)-苄氧氨基-哌啶-2-甲酸是制备阿维巴坦的关键中间体,因此开发一种操作简便、收率高、立体选择性好的方法合成(2s,5r)-苄氧氨基-哌啶-2-甲酸很有必要。
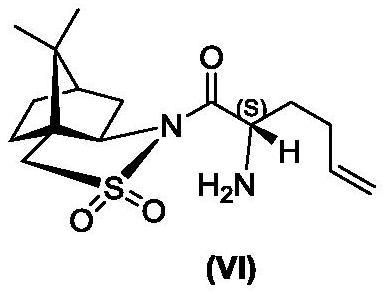
技术实现思路
1、本发明提供了一种以廉价易得的l-樟脑磺酰胺为手性源,经缩合、不对称烷基化、水解、脱手性辅助基制得阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸的方法。
2、阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸的制备方法,所述方法包括如下步骤:
3、
4、其中:式(iv)中,x选自cl、br或i。本发明采用如下技术方案:
5、一种阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸的制备方法,按照如下步骤进行:
6、步骤a:式(i)所示的l-樟脑磺酰胺溶于有机溶剂a1中,在保护氛围a(本发明的实施例中为氮气)中,10~30℃(优选20-25℃)条件下滴加三甲基铝的正己烷溶液,滴毕,搅拌反应10-30分钟,得到反应液a1;将式(ⅱ)所示的二苯基亚胺酯溶于有机溶剂a2,加入所述反应液a1中,在20~80℃(优选为40-50℃)下反应10-40小时(优选为15-20小时),反应结束后,所得反应液a经后处理a,得到式(ⅲ)所示化合物(具有手性);
7、所述的式(i)所示的l-樟脑磺酰胺、式(ⅱ)所示的二苯基亚胺酯与所述三甲基铝的正己烷溶液所含三甲基铝的物质的量之比为1.0:1.0-1.5:1.0-1.5(优选1.0:1.1:1.2);
8、
9、步骤b:步骤a中得到的式(ⅲ)所示的化合物溶于有机溶剂b中,在-78℃、保护氛围b下加入碱性物质b,然后在-78℃搅拌反应10-60分钟(优选20-30分钟),在-78℃加入式(iv)所示的高烯丙基卤化物,加毕,自然升至室温(20-25℃)反应3-8小时(优选为4-6小时),所得反应液b经后处理b得到式(v)所示的化合物;
10、所述的碱性物质b选自氢化钠、叔丁醇钾、叔丁醇钠、正丁基锂、叔丁基锂、二异丙基氨基锂、六甲基二硅基胺基钾、双(三甲基硅基)氨基钠、六甲基二硅基胺基锂之一,优选为六甲基二硅基胺基锂;
11、所述式(iii)所示的化合物、式(iv)所示的高烯丙基卤化物与碱性物质b的物质的量之比为1.0:1.0-1.5:1.0-1.5(优选1.0:1.2:1.2);
12、
13、式iv中,x为卤素,例如cl、br或i,在本发明的一个实施例中为氯;
14、步骤c:步骤b中得到的式(v)所示的化合物溶于有机溶剂c中,在-10℃~30℃(优选0℃~20℃)下加入酸性物质的水溶液反应1-8小时(优选为3-4小时),自然升至室温,用饱和碳酸氢钠溶液将反应液ph调至8-9,所得反应液c经后处理c得到式(vi)所示的化合物;这里加酸水解就能得到产物的盐酸盐,调ph主要是中和盐酸,得到游离氨基。
15、所述的式(v)所示的化合物与所述酸性物质的水溶液所含酸性物质的物质的量之比为1.0:1.0-2.0(优选为1.0:1.5);
16、
17、步骤d:步骤c中得到的式(vi)所示的化合物溶于有机溶剂d中,加入单质碘,在0℃-70℃(优选10℃-30℃)反应3-10小时(优选为5-7小时),所得反应液d经后处理d得到式(vii)所示化合物;
18、所述的式(vi)所示的化合物和单质碘的物质的量之比为1.0:1.0-2.0(优选为1.0:1.2);
19、
20、步骤e:步骤d中得到的式(vii)所示的化合物溶于有机溶剂e中,加入苄氧羟胺盐酸盐和有机碱e,在20℃-100℃(优选40℃-50℃)反应2-8小时(优选为3-5小时),得到反应液e经后处理e得到式(viii)所示化合物;
21、所述式(vii)所示的化合物、苄氧羟胺盐酸盐与有机碱e的物质的量之比为1.0:1.0-2.0:1.0-2.0(优选为1.0:1.5:1.5);
22、
23、步骤f:式(viii)所示的化合物溶于有机溶剂f中,在-10℃-30℃(优选0-20℃)下,加入碱性物质f反应1-10小时(优选为2-5小时),自然升至室温(20-25℃),加入盐酸(本发明的一个实施例中浓度为1.0mol/l)调节ph至4-5,得到反应液f经后处理f,得到式(ix)所示的阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸;
24、
25、所述的碱性物质f优选无机碱如氢氧化钠、氢氧化锂、氢氧化钾、氢氧化钙,最优选氢氧化锂,所述的碱性物质f以碱性物质f的水溶液的形式加入,在本发明的一个实施例中所述的碱性物质f的浓度为2.0mol/l;
26、所述式(viii)所示的化合物与碱性物质f的物质的量之比为1.0:1.0-5.0,优选1.0:3.0。。
27、进一步,步骤a中,所述的有机溶剂a1、有机溶剂a2各自独立为四氢呋喃、2-甲基四氢呋喃、甲苯、二甲基亚砜中的一种或两种以上的混合溶剂,优选为甲苯。
28、进一步,步骤a中,所述的有机溶剂a1的体积以所述的式(i)所示的l-樟脑磺酰胺的质量计为10-15ml/g;所述的有机溶剂a2的体积以所述的式(ⅱ)所示的二苯基亚胺酯的质量计为5-10ml/g。
29、进一步,在本发明的实施例中,所述三甲基铝的正己烷溶液的浓度为1.0mol/l-2.0mol/l。
30、进一步,步骤a中,所述后处理a为:将所述反应液a降至室温,依次加入饱和碳酸氢钠溶液、水,分液,水相用乙酸乙酯萃取,合并有机相,水洗(三次),浓缩有机相,以体积比为4:1的石油醚和乙酸乙酯的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得到式(iii)所示的化合物。
31、进一步,步骤b中,所述的有机溶剂b为甲苯、无水四氢呋喃、1,4-二氧六环中的一种或两种以上的混合溶剂,优选为无水四氢呋喃。
32、进一步,步骤b中,所述的有机溶剂b的体积以所述的式(iii)所示的化合物的质量计为5-10ml/g。
33、进一步,步骤b中,所述后处理b为:向所述反应液b中加入水,分液,所得水相用乙酸乙酯萃取(3次),合并有机相,用饱和氯化铵洗涤,所得有机相浓缩,以体积比为5:1的石油醚和乙酸乙酯的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得到式(v)所示的化合物。
34、进一步,步骤c中,所述的酸性物质优选为氯化氢或硫酸,最优选氯化氢。
35、更进一步,在本发明的一个实施例中,所述酸性物质以酸性物质的水溶液形式加入,所述酸性物质的水溶液的浓度为1.0mol/l。
36、进一步,步骤c中,所述的有机溶剂c为甲醇、四氢呋喃、n,n-二甲基甲酰胺中的一种或两种以上的混合溶剂,优选四氢呋喃。
37、进一步,步骤c中,所述的有机溶剂c的体积以所述的式(v)所示的化合物的质量计为10-15ml/g。
38、进一步,步骤c中,所述后处理c为:所述反应液c用乙酸乙酯萃取,有机相浓缩,以体积比为1:1的石油醚和乙酸乙酯的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得到式(vi)所示的化合物。
39、进一步,步骤d中,所述的有机溶剂d为二氯甲烷、氯仿、1,2-二氯乙烷、甲苯中的一种或两种以上的混合溶剂,优选为二氯甲烷。
40、进一步,步骤d中,所述的有机溶剂d的体积以所述的式(vi)所示的化合物的质量计为10-15ml/g。
41、进一步,步骤d中,所述后处理d为:将所述反应液d中加入饱和亚硫酸氢钠水溶液,乙酸乙酯萃取,所得有机相减压浓缩,以体积比为2:1的石油醚和乙酸乙酯的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得到式(vii)所示的化合物。
42、进一步,步骤e中,所述的有机溶剂e为二氯甲烷、氯仿、1,2-二氯乙烷,甲苯、四氢呋喃、乙酸乙酯中的一种或两种以上的混合溶剂,优选为1,2-二氯乙烷。
43、进一步,步骤e中,所述的有机溶剂e的体积以所述的式(vii)所示的化合物的质量计为10-15ml/g。
44、进一步,步骤e中,所述的有机碱e选自三乙胺、二异丙基乙基胺、吡啶、2,6-二甲基吡啶中的一种或两种的混合物,优选三乙胺。
45、进一步,步骤e中,所述后处理e为:向所述反应液e中加入水,再用乙酸乙酯萃取,所得有机相浓缩,以体积比为1:1的石油醚和乙酸乙酯的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得到式(viii)所示的化合物。
46、进一步,所述的碱性物质f以碱性物质f的水溶液的形式加入,所述的碱性物质f的水溶液的浓度为2.0mol/l。
47、进一步,步骤f中,所述的碱性物质f选自氢氧化钠、氢氧化锂、氢氧化钾、氢氧化钙中的一种或两种的混合物,优选氢氧化锂。
48、进一步,步骤f中,所述的有机溶剂f为甲醇、乙醇、1,4-二氧六环、四氢呋喃中的一种或两种以上的混合溶剂,优选四氢呋喃。
49、进一步,步骤f中,所述的有机溶剂f的体积以所述的式(vi)所示的化合物的质量计为10-15ml/g。
50、进一步,步骤f中,所述的反应液f的后处理f的方法为:所得反应液f减压蒸馏除去溶剂,以体积比为1:1的二氯甲烷与甲醇的混合液为洗脱剂进行柱层析分离,收集含目标产物的洗脱液,蒸除溶剂得式(ix)所示的阿维巴坦中间体(2s,5r)-苄氧氨基-哌啶-2-甲酸。
51、与现有技术相比,本发明的有益效果在于:
52、本发明所述的原料价廉易得、路线简短、收率高、立体选择性好,式(ii)所示的二苯基亚胺酯与三甲基铝形成络合物,可以提高樟脑磺内酰胺中氨基的亲核能力,减少副产物的产生。目前(2s,5r)-苄氧氨基-哌啶-2-甲酸为阿维巴坦的重要手性中间体,具有良好的市场前景。
- 还没有人留言评论。精彩留言会获得点赞!