用于治疗认知损害的苯并二氮杂环庚三烯衍生物,组合物和方法与流程
发明领域本发明涉及化合物、组合物和方法,其用于在有需要的受试者中治疗与中枢神经系统(cns)障碍有关的认知损害,与脑癌有关的认知损害,和脑癌。
背景技术:
0、发明背景
1、作为衰老的正常后果或作为中枢神经障碍的后果,认知能力可以减退。
2、例如,大部分老龄人会经历认知能力的减退,其超过在正常衰老中典型的认知能力的减退。认知功能的这类年龄相关的丧失在临床上的特征在于记忆,认知,推理和判断的进行性丧失。轻度认知损害(mci),年龄相关的记忆损害(aami),年龄相关的认知减退(arcd)或类似的临床分组是与这类年龄相关的认知功能丧失相关的病症之一。根据一些估计,仅美国就有超过1,600万人患有aami(barker等人,1995),并且据估计mci会影响美国的550-700万65岁以上的人(plassman等人,2008)。
3、认知损害也与其它中枢神经系统(cns)障碍相关,诸如痴呆,阿尔茨海默病(ad),前驱症状的ad(prodromal ad),创伤后应激障碍(ptsd),精神分裂症,双相型障碍(特别是躁狂症),肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍(autism spectrum disorder),脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。
4、因此,存在对有效治疗与中枢神经系统(cns)障碍相关的认知损害和改善诊断为患有例如年龄相关的认知损害,mci,遗忘性mci,aami,arcd,痴呆,ad,前驱症状的ad,ptsd,精神分裂症或双相型障碍(特别是躁狂症),肌萎缩侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾和伴认知损害的类似中枢神经系统(cns)障碍或处于发展它们的风险中的患者的认知功能的需要。
5、gabaa受体(gabaa r)是来自不同亚基(α1-6,β1-3,γ1-3,δ,ε,π,θ)库的五聚体装配物,所述不同亚基形成由神经递质γ-氨基丁酸(gaba)门控的cl-可透过的通道。不同的药理学效应(包括焦虑症,癫痫症,失眠症,前驱麻醉镇静和肌松弛)由不同的gabaa亚型介导。
6、各种研究已经证明,减少的gaba信号传递与具有认知损害的不同cns障碍相关。具体地,在哺乳动物脑中相对稀少的含有α5的gabaa r在改变学习和记忆中起作用。在先的研究证明,随着年龄相关的认知衰退,大鼠中gabaa受体的α5亚基的海马表达减少(参见国际专利公开wo 2007/019312)。这样的结果提示,含有α5的gabaa r功能的上调可有效地治疗与所述cns障碍相关的认知损害。
7、因此,存在对可用于治疗与所述cns障碍相关的认知损害的治疗制剂的含有α5的gabaa r的正变构调节剂的需要。
技术实现思路
1、本发明通过提供式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合解决了上述需要:
2、
3、其中:
4、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
5、a是c,cr6或n;
6、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
7、d是n,nr7,o,cr6或c(r6)2;
8、e是n,nr7,cr6或c(r6)2;
9、w是n,nr7,cr6或c(r6)2;
10、x是n,nr7,o,cr6或c(r6)2;
11、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
12、v是c或cr6,
13、或者当z是c或cr6时,v是c,cr6或n;
14、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10;且其中r2独立地被0-5个r’取代;
15、m和n独立地是选自0-4的整数;
16、p是选自2-4的整数;
17、键的每次出现是单键或双键;
18、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,
19、
20、-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
21、r3不存在或选自:
22、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
23、每个r6独立地是-h或-(c1-c6)烷基;
24、每个r7独立地是-h或-(c1-c6)烷基;
25、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
26、每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;
27、每个r独立地选自:
28、h-,
29、(c1-c12)-脂族基-,
30、(c3-c10)-环烷基-,
31、(c3-c10)-环烯基-,
32、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
33、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
34、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
35、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
36、(c6-c10)-芳基-,
37、(c6-c10)-芳基-(c1-c12)脂族基-,
38、(c6-c10)-芳基-o-(c1-c12)脂族基-,
39、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
40、3-10元杂环基-,
41、(3-10元杂环基)-(c1-c12)脂族基-,
42、(3-10元杂环基)-o-(c1-c12)脂族基-,
43、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
44、5-10元杂芳基-,
45、(5-10元杂芳基)-(c1-c12)-脂族基-,
46、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
47、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
48、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
49、其中r的每次出现独立地被0-5个r’取代;
50、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
51、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
52、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2nro2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
53、本技术的某些实施方案提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
54、
55、其中:
56、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
57、a是c,cr6或n;
58、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
59、d是n,nr7,o,cr6或c(r6)2;
60、e是n,nr7,cr6或c(r6)2;
61、w是n,nr7,cr6或c(r6)2;
62、x是n,nr7,o,cr6或c(r6)2;
63、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
64、v是c或cr6,
65、或者当z是c或cr6时,v是c,cr6或n;
66、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10;且其中r2独立地被0-5个r’取代;
67、m和n独立地是选自0-4的整数;
68、p是选自2-4的整数;
69、键的每次出现是单键或双键;
70、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
71、r3不存在或选自:
72、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,
73、
74、-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
75、每个r6独立地是-h或-(c1-c6)烷基;
76、每个r7独立地是-h或-(c1-c6)烷基;
77、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
78、每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;
79、每个r独立地选自:
80、h-,
81、(c1-c12)-脂族基-,
82、(c3-c10)-环烷基-,
83、(c3-c10)-环烯基-,
84、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
85、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
86、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
87、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
88、(c6-c10)-芳基-,
89、(c6-c10)-芳基-(c1-c12)脂族基-,
90、(c6-c10)-芳基-o-(c1-c12)脂族基-,
91、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
92、3-10元杂环基-,
93、(3-10元杂环基)-(c1-c12)脂族基-,
94、(3-10元杂环基)-o-(c1-c12)脂族基-,
95、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
96、5-10元杂芳基-,
97、(5-10元杂芳基)-(c1-c12)-脂族基-,
98、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
99、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
100、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
101、其中r的每次出现独立地被0-5个r’取代;
102、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
103、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
104、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
105、本技术的某些实施方案提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
106、
107、其中:
108、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
109、a是c,cr6或n;
110、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
111、d是n,nr7,o,cr6或c(r6)2;
112、e是n,nr7,cr6或c(r6)2;
113、w是n,nr7,cr6或c(r6)2;
114、x是n,nr7,o,cr6或c(r6)2;
115、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
116、v是c或cr6,
117、或者当z是c或cr6时,v是c,cr6或n;
118、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8或-(ch2)nor8;
119、m和n各自独立地是选自0-4的整数;
120、键的每次出现是单键或双键;
121、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
122、r3不存在或选自:
123、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
124、每个r6独立地是-h或-(c1-c6)烷基;
125、每个r7独立地是-h或-(c1-c6)烷基;
126、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,-(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
127、每个r独立地选自:
128、h-,
129、(c1-c12)-脂族基-,
130、(c3-c10)-环烷基-,
131、(c3-c10)-环烯基-,
132、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
133、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
134、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
135、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
136、(c6-c10)-芳基-,
137、(c6-c10)-芳基-(c1-c12)脂族基-,
138、(c6-c10)-芳基-o-(c1-c12)脂族基-,
139、3-10元杂环基-,
140、(3-10元杂环基)-(c1-c12)脂族基-,
141、(3-10元杂环基)-o-(c1-c12)脂族基-,
142、5-10元杂芳基-,
143、(5-10元杂芳基)-(c1-c12)-脂族基-和
144、(5-10元杂芳基)-o-(c1-c12)-脂族基-;
145、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
146、其中r的每次出现独立地被0-5个r’取代;
147、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
148、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
149、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
150、在另一个方面,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
151、
152、其中m,r1,r2,r3,r4,r5和r6如在式i中所定义。
153、在另一个方面,本发明提供了式iii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
154、
155、其中m,r1,r2,r3,r4,r5和r6如在式i中所定义。
156、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
157、
158、其中r2是-or8,-sr8或-(ch2)nor8,其中r2独立地被0-5个r’取代,且其中m,n,r1,r3,r4,r5,r6和r8如在式i中所定义。
159、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
160、
161、其中r2是-(ch2)no(ch2)nr8,-(ch2)pr8或-(ch2)nn(r”)r10,其中r2独立地被0-5个r’取代,且其中m,n,p,r1,r3,r4,r5,r6,r8,r10和r”如本文中定义。
162、在又一方面,本发明提供式v化合物:
163、
164、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
165、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
166、a是c,cr6或n;
167、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
168、d是n,nr7,o,cr6或c(r6)2;
169、e是n,nr7,cr6或c(r6)2;
170、w是n,nr7,cr6或c(r6)2;
171、x是n,nr7,o,cr6或c(r6)2;
172、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
173、v是c或cr6,
174、或者当z是c或cr6时,v是c,cr6或n;
175、其中当由x,y,z,v和w形成的环是那么r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10;且其中r2独立地被0-5个r’取代;
176、m和n独立地是选自0-4的整数;
177、p是选自2-4的整数;
178、键的每次出现是单键或双键;
179、r1,r2,r4,和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)1-3-o(cr2)1-3-r,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,
180、-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2,-p(o)(h)(or),c≡c-r8,ch2cf3,和chf3;
181、r8的每次出现是-h,-(c1-c6)烷基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,-(c6-c10)芳基,-5-10元杂芳基,或-(c1-c6)烷基-5-10元杂芳基;
182、其中除了-h和-(c1-c6)烷基以外的每个r8独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
183、r3不存在或选自:
184、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2,-p(o)(h)(or),c≡c-r9,coome,cooet,-(c1-c6)烷基-c≡c-r10,ch2-or10,和ch2-o-ch2-r10;
185、其中每个r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-c(o)-(c6-c10)芳基,
186、
187、其中每个r9独立地被0-5个r11取代;
188、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基和-5-10元杂芳基,
189、其中r10选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c3-c6)环烷基,-ch2-(c3-c6)环烷基,-ch2-(c6-c10)芳基,和-ch2-5-10-元杂芳基,
190、其中每个r10独立地被0-5个r’取代;
191、其中r7选自-(c1-c6)烷基,-(c3-c6)环烷基,-5-10元杂芳基,-(c6-c10)芳基,-(c6-c10)芳基-(c1-c6)烷基,和-5-10元杂芳基-(c1-c6)烷基,和-5-10元杂芳基,
192、其中每个r7独立地被0-5个r’取代;
193、每个r6独立地是-h或-(c1-c6)烷基;
194、每个r7独立地是-h或-(c1-c6)烷基;
195、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基,或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
196、每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基,或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;
197、每个r独立地选自:
198、h-,
199、(c1-c12)-脂族基-,
200、(c3-c10)-环烷基-,
201、(c3-c10)-环烯基-,
202、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
203、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
204、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
205、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
206、(c6-c10)-芳基-,
207、(c6-c10)-芳基-(c1-c12)脂族基-,
208、(c6-c10)-芳基-o-(c1-c12)脂族基-,
209、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
210、3-10元杂环基-,
211、(3-10元杂环基)-(c1-c12)脂族基-,
212、(3-10元杂环基)-o-(c1-c12)脂族基-,
213、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
214、5-10元杂芳基-,
215、(5-10元杂芳基)-(c1-c12)-脂族基-,
216、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
217、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
218、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,和所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
219、其中r的每次出现独立地被0-5个r’取代;
220、或者两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
221、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
222、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-,和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。
223、在又一方面,本发明提供式vi化合物:
224、
225、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
226、m是0-3;
227、每个r1独立地选自:-卤素,-ome,-c≡c-r8,-cn,-chf2,-ch2cf3,-cf3,-ocf3,-(c1-c6)烷基,-(c6-c10)芳基,-(c1-c6)烷基-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-5-10元杂芳基,和-(c3-c6)环烷基;
228、其中r8是-h,-(c1-c6)烷基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,-(c6-c10)芳基,-5-10元杂芳基,或-(c1-c6)烷基-5-10元杂芳基;
229、其中除了-h和-(c1-c6)烷基以外的每个r8独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
230、r2是-卤素,-(cr2)1-3-or,-(cr2)1-3-o(cr2)1-3-r,-h,-(c1-c6)烷基,-(c6-c10)芳基,(c6-c10)芳基-(c1-c6)烷基-,-5-10元杂芳基,5-10元杂芳基-(c1-c6)烷基-,或-or9;
231、其中r的每次出现独立地选自-h,-(c1-c6)烷基,(c6-c10)芳基-,-5-10元杂芳基,(c6-c10)-芳基-(c1-c12)脂族基-,5-10元杂芳基-(c6-c10)烷基-,或-(c3-c6)环烷基;
232、其中除了-h和-(c1-c6)烷基以外的每个r独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代,
233、其中r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,和-(c1-c6)烷基-(c3-c6)环烷基;
234、其中每个r9独立地被0-5个r11取代;
235、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
236、r3选自:-卤素,-cn,-c≡cr9,coome,-cooet,-(c1-c6)烷基-c≡c-r10,-ch2-o-r10,-ch2-o-ch2-r10
237、
238、其中r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,和-c(o)-(c6-c10)芳基;
239、其中每个r9独立地被0-5个r11取代;
240、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
241、其中r10选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c3-c6)环烷基,-ch2-(c3-c6)环烷基,-ch2-(c6-c10)芳基,和-ch2-5-10-元杂芳基,
242、其中每个r10独立地被0-5个r’取代;
243、其中r7选自-(c1-c6)烷基,-(c3-c6)环烷基,-5-10元杂芳基,-(c6-c10)芳基,-(c6-c10)芳基-(c1-c6)烷基,和-5-10元杂芳基-(c1-c6)烷基,和-5-10元杂芳基;其中每个r7独立地被0-5个r’取代;
244、其中r3被0-5个r’取代;
245、r4和r5的每次出现独立地是-h,-(c1-c6)烷基或-(c1-c6)烷基-(c6-c10)芳基;所述(c6-c10)芳基独立地被0-5个-卤素取代;
246、每个r6独立地是-h或-(c1-c6)烷基;
247、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
248、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-,或(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基。
249、在又一方面,本发明提供式vii化合物:
250、
251、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
252、m是0-3;
253、每个r1独立地选自:-卤素,-ome,-c≡c-r9,-cn,-chf2,-ch2cf3,-cf3,-ocf3,-(c1-c6)烷基,-(c6-c10)芳基,-(c1-c6)烷基-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-5-10元杂芳基,和-(c3-c6)环烷基;
254、其中r9是-h,-(c1-c6)烷基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,-(c6-c10)芳基,-5-10元杂芳基,或-(c1-c6)烷基-5-10元杂芳基;
255、其中除了-h和-(c1-c6)烷基以外的每个r9独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
256、r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是-h,-(c1-c6)烷基,-(c6-c10)-芳基,5-10元杂芳基-,5-10元杂芳基-(c1-c6)烷基-,-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,或-(c1-c6)烷基-(c3-c6)环烷基;
257、其中除了-h和-(c1-c6)烷基以外的每个r8独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
258、其中n是0-4的整数;
259、其中r2独立地被0-5个r’取代;
260、r3选自:-卤素,-cn,-c≡cr9,coome,-cooet,-(c1-c6)烷基-c≡c-r10,-ch2-o-r10,-ch2-o-ch2-r10
261、
262、其中r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,和-c(o)-(c6-c10)芳基;
263、其中每个r9独立地被0-5个r11取代;
264、其中r10选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c3-c6)环烷基,-ch2-(c3-c6)环烷基,-ch2-(c6-c10)芳基,和-ch2-5-10-元杂芳基,
265、其中每个r10独立地被0-5个r’取代;
266、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
267、其中r7选自-(c1-c6)烷基,-(c3-c6)环烷基,-5-10元杂芳基,-(c6-c10)芳基,-(c6-c10)芳基-(c1-c6)烷基,和-5-10元杂芳基-(c1-c6)烷基,和-5-10元杂芳基;
268、其中每个r7独立地被0-5个r’取代;
269、其中r3被0-5个r’取代;
270、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
271、每个r6独立地是-h或-(c1-c6)烷基;
272、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2,-ome;
273、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-,和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个rt取代,所述rt独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。
274、在又一方面,本发明提供式viii化合物:
275、
276、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
277、m是0-3;
278、每个r1独立地选自:-卤素,-ome,-c≡c-r8,-chf2,-cf3,-ocf3,
279、其中r8是-h,-(c1-c6)烷基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,-(c6-c10)芳基,-5-10元杂芳基,或-(c1-c6)烷基-5-10元杂芳基;
280、其中除了-h和-(c1-c6)烷基以外的每个r8独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
281、r2是-h,-ch2-or,ch3,ch2-苯基;
282、其中r的每次出现独立地选自-(c1-c6)烷基,(c6-c10)芳基-,-5-10元杂芳基,(c6-c10)-芳基-(c1-c12)脂族基-,5-10元杂芳基-(c6-c10)烷基-,或-(c3-c6)环烷基;
283、其中除了-h和-(c1-c6)烷基以外的每个r独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代,
284、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
285、r3选自:-c≡cr9,-(c1-c6)烷基-c≡c-r10,-ch2-o-r10,
286、
287、其中r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,和-c(o)-(c6-c10)芳基;
288、其中每个r9独立地被0-5个r11取代;
289、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
290、其中r10选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c3-c6)环烷基,-ch2-(c3-c6)环烷基,-ch2-(c6-c10)芳基,和-ch2-5-10-元杂芳基,
291、其中每个r10独立地被0-5个r’取代;
292、其中r7选自-(c1-c6)烷基,-(c3-c6)环烷基,-5-10元杂芳基,-(c6-c10)芳基,-(c6-c10)芳基-(c1-c6)烷基,和-5-10元杂芳基-(c1-c6)烷基,和-5-10元杂芳基;其中每个r7独立地被0-5个r’取代;
293、r4和r5的每次出现独立地是-h,-(c1-c6)烷基或-(c1-c6)烷基-(c6-c10)芳基;所述(c6-c10)芳基独立地被0-5个-卤素取代;
294、每个r6独立地是-h或-(c1-c6)烷基。
295、在又一方面,本发明提供式ix化合物:
296、
297、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
298、每个r1独立地选自:-cl,-ome,-c≡c-r9,-chf2,-cf3,和-ocf3;
299、其中r9是-h,-(c1-c6)烷基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,-(c6-c10)芳基,-5-10元杂芳基,或-(c1-c6)烷基-5-10元杂芳基;
300、其中除了-h和-(c1-c6)烷基以外的每个r9独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
301、r2是-h,ch2or8,ch3,ch2-苯基,其中r8的每次出现独立地是-h,-(c1-c6)烷基,-(c6-c10)-芳基,5-10元杂芳基-,5-10元杂芳基-(c1-c6)烷基-,-(c3-c6)环烷基,-(c1-c6)烷基-(c6-c10)芳基,或-(c1-c6)烷基-(c3-c6)环烷基;
302、其中除了-h和-(c1-c6)烷基以外的每个r8独立地被-卤素,-(c1-c6)烷基,-cf3,-ocf3或o-(c1-c6)烷基中的0-5个取代;
303、r3选自:-c≡cr9,-(c1-c6)烷基-c≡c-r10,
304、
305、其中r9选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c1-c6)烷基-(c6-c10)芳基,-(c1-c6)烷基-5-10元杂芳基,-(c3-c6)环烷基,-(c1-c6)烷基-(c3-c6)环烷基,和-c(o)-(c6-c10)芳基;
306、其中每个r9独立地被0-5个r11取代;
307、其中r10选自-h,-(c1-c6)烷基,-(c6-c10)芳基,-5-10元杂芳基,-(c3-c6)环烷基,-ch2-(c3-c6)环烷基,-ch2-(c6-c10)芳基,和-ch2-5-10-元杂芳基,
308、其中每个r10独立地被0-5个r’取代;
309、其中r11的每次出现独立地选自-卤素,-cf3,-ocf3,-ome,-(c6-c10)芳基,-(c1-c6)烷基,和-5-10元杂芳基,
310、其中r7选自-(c1-c6)烷基,-(c3-c6)环烷基,-5-10元杂芳基,-(c6-c10)芳基,-(c6-c10)芳基-(c1-c6)烷基,和-5-10元杂芳基-(c1-c6)烷基,和-5-10元杂芳基;
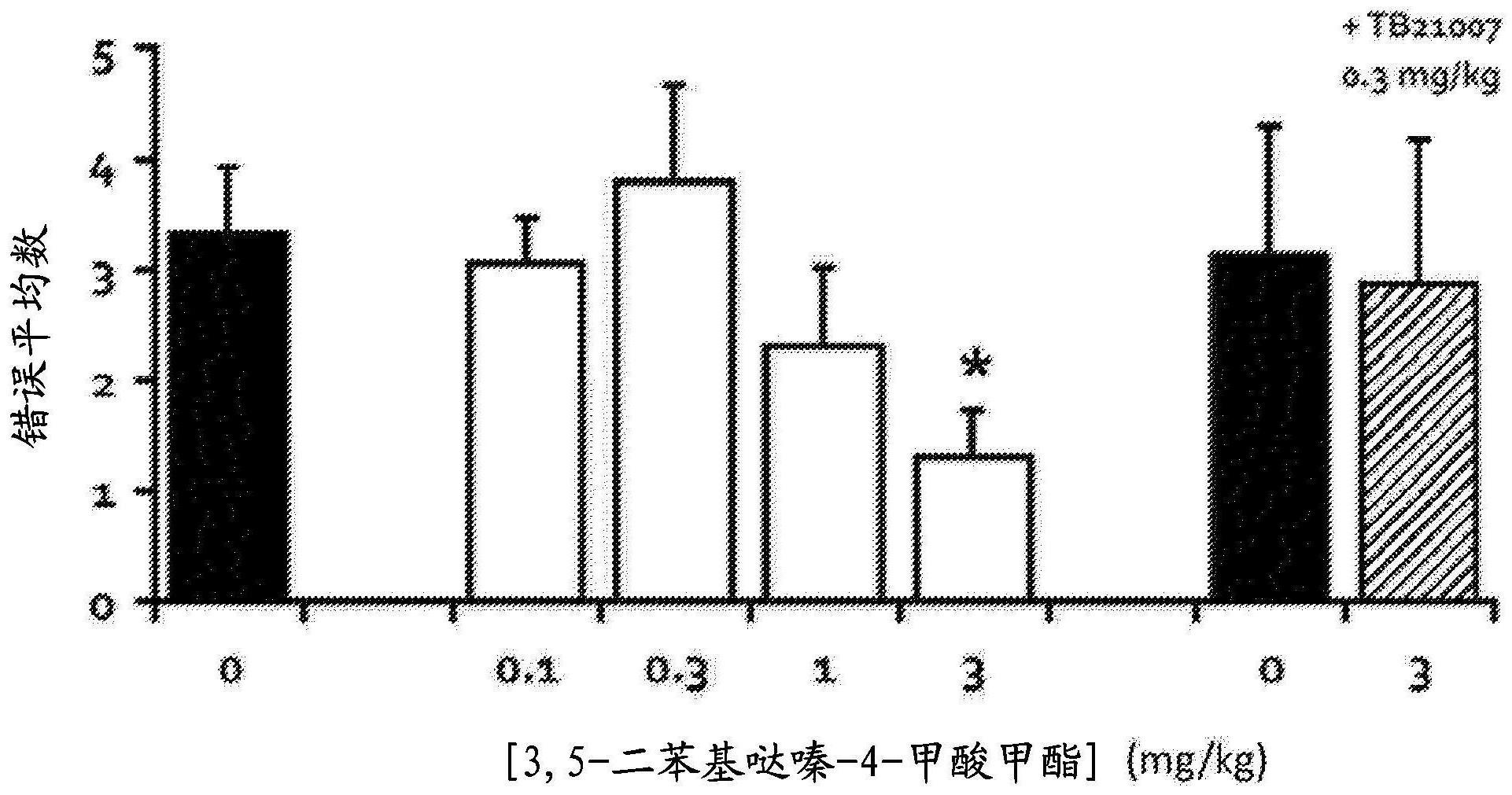
311、其中每个r7独立地被0-5个r’取代;
312、其中r3被0-5个r’取代;
313、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
314、每个r6独立地是-h或-(c1-c6)烷基。
315、本发明还提供了药物组合物,其包含式i,ii,iii,iv,v,vi,vii,viii或ix的化合物或药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或它们的组合。
316、在某些实施方案中,式i的化合物是gabaaα5受体正变构调节剂。在某些实施方案中,式ii的化合物是gabaaα5受体正变构调节剂。在某些实施方案中,式iii的化合物是gabaaα5受体正变构调节剂。在某些实施方案中,式iv的化合物是gabaaα5受体正变构调节剂。在某些实施方案中,式v的化合物是gabaaα5受体正变构调节剂。在某些实施方案中,式vi的化合物是gabaaα5受体正变构调节剂。式i,ii,iii,iv,v,vi,vii,viii或ix的化合物可以用于治疗本文所述的病症,诸如通过作为gabaaα5受体正变构调节剂的活性。
317、在本发明的另一个方面,提供了在需要治疗或处于与cns障碍相关的认知损害风险中的受试者中治疗与cns障碍相关的认知损害的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在某些实施方案中,所述具有认知损害的cns障碍包括但不限于,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。在本发明的另一个方面,提供了在有此需要的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在本发明的某些实施方案中,每12或24小时施用本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
318、在本发明的又一方面,提供用于治疗脑癌(包括脑肿瘤例如成神经管细胞瘤)的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在本发明的又一方面,提供在患脑癌(包括脑肿瘤例如成神经管细胞瘤)的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在本发明的某些实施方式中,每12或24小时施用本发明化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
319、在某些实施方案中,本发明的化合物和组合物用于用作药物。在某些实施方案中,本发明的化合物和组合物用于在需要治疗或处于与cns障碍相关的认知损害风险中的受试者中治疗与cns障碍相关的认知损害。在某些实施方案中,所述具有认知损害的cns障碍包括但不限于,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。在某些实施方式中,本发明化合物和组合物用作治疗脑癌(包括脑肿瘤例如成神经管细胞瘤)的药物。在某些实施方式中,本发明化合物和组合物用作治疗与脑癌(包括脑肿瘤例如成神经管细胞瘤)有关的认知损害的药物。
320、在某些实施方案中,本技术提供了本文描述的化合物或组合物在药物制备中的用途,所述药物用于在需要治疗或处于与cns障碍相关的认知损害风险中的受试者中治疗与cns障碍相关的认知损害。在某些实施方案中,所述具有认知损害的cns障碍包括但不限于,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。在某些实施方式中,本发明化合物和组合物用于制备药物,所述药物用于治疗脑癌(包括脑肿瘤例如成神经管细胞瘤)。在某些实施方式中,本发明化合物和组合物用于制备药物,所述药物用于治疗与脑癌(包括脑肿瘤例如成神经管细胞瘤)有关的认知损害。
321、附图详述
322、图1是描述在8-臂放射臂迷宫(ram)试验中给10只老龄受损(ai)大鼠施用3,5-二苯基哒嗪-4-甲酸甲酯对空间记忆保留的影响的图。黑色条表示用单独的媒介物治疗的大鼠;空心条表示用不同剂量的3,5-二苯基哒嗪-4-甲酸甲酯治疗的大鼠;带阴影线的条表示用tb21007和3,5-二苯基哒嗪-4-甲酸甲酯的组合治疗的大鼠。
323、图2是显示3,5-二苯基哒嗪-4-甲酸甲酯(静脉内地施用)对海马和小脑中ro154513的结合的影响的图。3,5-二苯基哒嗪-4-甲酸甲酯阻断海马中ro154513的结合,但不影响在小脑中ro15413的结合。
324、图3是显示静脉内施用的3,5-二苯基哒嗪-4-甲酸甲酯的剂量依赖性gabaaα5受体占有率的图,其中根据ro 15-4513的海马(高gabaaα5受体密度区域)暴露与ro 15-4513的小脑(具有低gabaaα5受体密度的区域)暴露之比率或通过使用gabaaα5选择性化合物l-655,708(10mg/kg,静脉内)确定完全占有率来确定受体占有率。
325、图4是显示海马中3,5-二苯基哒嗪-4-甲酸甲酯的暴露占有率相关性的图。3,5-二苯基哒嗪-4-甲酸甲酯在老龄受损大鼠中具有行为活性的暴露时的gabaaα5的受体占有率约为32%。
326、图5是描述在8-臂放射臂迷宫(ram)试验中3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯对10只老龄受损(ai)大鼠的空间记忆保留的影响的图。图5显示3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯在ram试验中对10只老龄受损(ai)大鼠的空间记忆保留的影响,其中将媒介物对照品测试3次,将不同剂量的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯测试2次;在图5中,黑色条表示用单独的媒介物治疗的大鼠,空心条表示用不同剂量的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯治疗的大鼠。
327、图6是显示3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯(静脉内地施用)对海马和小脑中ro154513的结合的影响的图。3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯阻断海马中的ro154513的结合,但不影响小脑中ro15413的结合。
328、图7是显示静脉内施用的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯的剂量依赖性gabaaα5受体占有率的图,正如根据ro 15-4513的海马(高gabaaα5受体密度区域)暴露与ro 15-4513的小脑(具有低gabaaα5受体密度的区域)暴露之比确定完全占有率计算的。
329、图8(a)-(c)是使用莫里斯(morris)水迷宫行为任务显示6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮与媒介物二甲亚砜(dmso)相比在老龄受损大鼠中的影响的图。图8(a)显示接受6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮的大鼠和接受媒介物dmso的大鼠在训练过程中的逃逸潜伏期(即大鼠在水池中找到隐藏平台所花费的以秒计的平均时间);图8(b)显示接受6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮的大鼠和接受媒介物dmso的大鼠在目标环形区域(annulus)和相对的环形区域中花费的时间量;图8(c)显示接受6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮的大鼠和接受媒介物dmso的大鼠在目标环形区域和相对的环形区域中交叉通过的次数。
330、发明详述
331、定义
332、除非本文另有定义,否则本技术中使用的科学和技术术语应具有本领域技术人员通常所理解的含义。一般而言,本文所述的化学,细胞和组织培养,分子生物学,细胞和癌症生物学,神经生物学,神经化学,病毒学,免疫学,微生物学,药理学,遗传学和蛋白质和核酸化学的技术和与之关联使用的命名法是本领域中众所周知且普遍使用的那些。
333、除非另外指明,否则本发明的方法和技术通常根据本领域众所周知的常规方法和如贯穿本说明书引述和讨论的各种一般和更具体的参考文献中所述进行。参见,例如"principles of neural science",mcgraw-hill medical,new york,n.y.(2000);motulsky,"intuitive biostatistics",oxford university press,inc.(1995);lodish等人,"molecular cell biology,第4版",w.h.freeman&co.,new york(2000);griffiths等人,"introduction to genetic analysis,第7版",w.h.freeman&co.,n.y.(1999);gilbert等人,"developmental biology,第6版",sinauer associates,inc.,sunderland,ma(2000)。
334、根据本领域的常规应用使用如本文使用的化学术语,以"the mcgraw-hilldictionary of chemical terms",parker s.,编,mcgraw-hill,san francisco,c.a.(1985)中为典型。
335、特别将本技术中涉及的所有公开文献,专利和公布的专利申请通过引用并入本文。在冲突的情况下,以本说明书包括其具体定义为准。
336、贯穿本说明书,词语"包含"或变体诸如"包括"或"含有"应理解为暗示包括所述的整数(或组分)或整数(或组分)的组,但是不排除任何其它整数(或组分)或整数(或组分)的组。
337、除非上下文另外清楚地指明,否则单数形式"一个","一种"和"所述"包括复数形式。
338、术语"包括"用于指"包括但不限于"。"包括"和"包括但不限于"互换使用。
339、术语"试剂"在本文中用于表示化学化合物(诸如有机或无机化合物(包括例如本发明的化合物),化学化合物的混合物),生物大分子(诸如核酸,抗体(包括其部分)以及人源化,嵌合和人抗体和单克隆抗体,蛋白质或其部分,例如肽,脂质,碳水化合物)或由生物材料诸如细菌,植物,真菌或动物(特别是哺乳动物)细胞或组织制成的提取物。试剂包括,例如就结构而言已知的试剂和就结构而言未知的试剂。这样的试剂的含有α5的gabaa受体激动剂活性可以使它们适合作为本发明的方法和组合物中的"治疗剂"。
340、"患者","受试者"或"个体"互换使用,且表示人或非人动物。这些术语包括哺乳动物,诸如人类,灵长类动物,家畜动物(包括牛,猪等),伴侣动物(例如,犬科动物,猫科动物等)和啮齿类动物(例如,小鼠和大鼠)。
341、"认知功能"或"认知状态"表示分别涉及学习和/或记忆的任何高级智力脑过程或脑状态,包括但不限于注意,信息采集,信息加工,工作记忆,短期记忆,长期记忆,顺应性记忆,逆行性记忆,记忆恢复,辨别学习,决策制定,抑制响应控制,注意设定-移位,延迟强化学习,逆向学习,自发行为的瞬时整合,对某环境和自我护理表示关注,处理速度,推理和问题解决和社会认知。
342、在人类中,可测定认知功能,例如,但不限于,通过临床总体印象的变化量表(clinical global impression of change scale)(cibic-plus scale);简短精神状态检查(the mini mental state exam)(mmse);神经精神调查(the neuropsychiatricinventory)(npi);临床痴呆等级量表(the clinical dementia rating scale)(cdr);剑桥神经心理测试自动成套测试(the cambridge neuropsychological test automatedbattery)(cantab);老龄医学的桑多临床评价(the sandoz clinical assessment-geriatric)(scag),buschke选择联想测试(the buschke selective reminding test)(buschke和fuld,1974);口头配对相关分测试(the verbal paired associatessubtest);逻辑记忆分测试(the logical memory subtest);修订的韦克斯勒记忆量表(the wechsler memory scale-revised)(wms-r)的视觉复现分测试(the visualreproduction subtest)(wechsler,1997);本顿视觉保留试验(the benton visualretention test),或外显3选项强迫选择任务(the explicit 3-alternative forcedchoice task)或精神分裂症认知功能成套测试共识版(matrics consensusneuropsychological test battery)。参见folstein等人,jpsychiatric res 12:189-98,(1975);robbins等人,dementia 5:266-81,(1994);rey,l'examen clinique enpsychologie,(1964);kluger等人,j geriatr psychiatry neurol 12:168-79,(1999);marquis等人,2002以及masur等人,1994。另外参见buchanan,r.w.,keefe,r.s.e.,umbricht,d.,green,m.f.,laughren,t.和marder,s.r.(2011),the fda-nimh-matricsguidelines for clinical trial design of cognitive-enhancing drugs:what do weknow 5years later?schizophr.bull.37,1209-1217。
343、在动物模型系统中,可按照本领域已知的各种常规方式测定认知功能,包括使用其中动物使用空间信息的莫里斯水迷宫(mwm),巴恩斯环形迷宫,高架放射臂迷宫,t形迷宫或任何其它迷宫。可以通过逆向学习,度外定势转换(extradimensional set shifting),条件性辨别学习和奖赏期待的评定来评价认知功能。本领域已知的其它试验也可用于评价认知功能,诸如新物体识别和气味识别任务。
344、还可使用成像技术测定认知功能,诸如正电子发射断层摄影术(pet),功能性磁共振成像(fmri),单光子发射计算体层摄影(spect)或任何其它能够测定脑功能的成像技术。在动物中,还可使用电生理技术测定认知功能。
345、"促进"认知功能表示影响受损的认知功能,使得它更接近类似于正常,未受损的受试者的功能。可将认知功能促进至任何可检测的程度,但在人类中,优选将其促进至足以允许受损受试者以与正常未受损的受试者或年龄匹配的正常未受损受试者尽可能接近的熟练水平(level of proficiency)进行每日正常生活的活动。
346、在一些情况下,"促进"在受与年龄相关认知影响的受试者中的认知功能表示影响受损的认知功能,使得它更接近类似于年龄匹配的正常,未受损的受试者的功能或年轻成年受试者的功能。可将该受试者的认知功能促进至任何可检测的程度,但在人类中,优选将其促进至足以允许受损受试者以与正常未受损受试者或年轻成年受试者或年龄匹配正常未受损受试者尽可能接近的熟练水平进行每日正常生活的活动。
347、"保留"认知功能表示影响正常或受损认知功能,使得它不减退或不降至低于在首次呈现或诊断时在受试者中观察到的水平或延迟这种减退。
348、"改善"认知功能包括在受试者中促进认知功能和/或保留其认知功能。
349、"认知损害"表示在受试者中的认知功能并不如在正常,未受损受试者中所预期的一样强。在一些情况下,认知功能与在正常,未受损受试者中所预期的认知功能相比减少约5%,约10%,约30%,或更多。在一些情况下,在受与年龄相关认知损害影响的受试者中的"认知损害"表示在受试者中的认知功能并不象在年龄匹配的正常,未受损受试者中所预期的或年轻成年受试者(即,在认知测定中对于给定年龄具有平均得分的受试者)中所预期的功能一样强。
350、"年龄相关的认知损害"表示在老龄受试者中的认知损害,其中他们的认知功能并不如在年龄匹配的正常的受试者中所预期的或在年轻成年受试者中所预期的一样强。在一些情况下,认知功能与在年龄匹配的正常受试者中所预期的认知功能相比减少约5%,约10%,约30%,或更多。在一些情况下,认知功能是如在年龄匹配的正常受试者中所预期的,但与在年轻成年受试者中所预期的认知功能相比减少约5%,约10%,约30%,约50%,或更多。与年龄相关受损的认知功能可能与轻度认知损害(mci)(包括遗忘性mci和非遗忘性mci),年龄相关的记忆损害(aami)和年龄相关的认知减退(arcd)相关。
351、与ad相关的或涉及ad或在ad中的"认知损害"表示在受试者中的认知功能并不如在使用常规方法和标准未诊断出ad的受试者中所预期的一样强。
352、"轻度认知损害"或"mci"表示特征在于未伴随其它认知异常和相对正常功能的能力的单独的记忆损害的病症。mci的临床特征的一组标准指定了如下特征:(1)记忆抱怨(如患者,填报人或医师报道的);(2)正常日常生活活动(adl);(3)正常总体认知功能;(4)年龄记忆异常(定义为低于指定年龄平均值多于1.5标准偏差的得分);和(5)不存在痴呆指示(如根据dsm-iv指导原则定义)。petersen等人,srch.neurol.56:303-308(1999);petersen,"mild cognitive impairment:aging to alzheimer's disease."oxforduniversity press,n.y.(2003)。具有mci的受试者中的认知缺陷可以涉及任意的认知区域或心理过程,包括记忆,语言,相关性,注意力,感觉,问题解决,执行功能和视觉空间技能。参见,例如winbald等人,j.intern.med.256:240-240,2004;meguro,acta.neurol.taiwan.15:55-57,2008;ellison等人,cns spectr.13:66-72,2008,petersen,semin.neurol.27:22-31,2007。mci进一步被再分成遗忘型mci(amci)和非遗忘型mci,其特征在于特别地记忆损害(或其缺乏)。将mci定义为amci,条件是发现记忆因受试者年龄和受教育程度水平而受损。另一方面,如果发现受试者记忆因年龄和教育而完整,但其它非记忆认知域受损,诸如语言,执行功能或视觉空间技能,则mci被定义为非遗忘型mci。amci和非遗忘型mci都可以进一步被再分成单一或多域型mci。amci-单一域表示这样已知病症,其中记忆而非其它认知域受损。amci-多域表示这样一种病症,其中记忆和至少另一种认知域受损。非遗忘型mci是单一域或多域的,这取决于是否非一种以上非记忆认知域受损。参见,例如peterson和negash,cns spectr.13:45-53,2008。
353、mci诊断通常要求客观评价认知损害,可通过使用充分确立的神经心理学测验获取,包括简短精神状态检查(mmse),剑桥神经心理测试自动成套测试(cantab)和个体测试诸如rey听觉言语学习测试(avlt),修订的韦克斯勒记忆量表的逻辑记忆分测试(wms-r)和纽约大学(nyu)段落回忆测试(paragraph recall test)。参见folstein等人,jpsychiatric res 12:189-98(1975);robbins等人,dementia 5:266-81(1994);kluger等人,j geriatric psychiatry neurol 12:168-79(1999)。
354、"年龄相关的记忆损害(aami)"表示因老龄化导致的记忆减退。如果他或她至少为50岁且满足如下全部标准,则可将患者视为具有aami:a)患者注意到记忆表现减退;b)与年轻成年人相比患者在记忆标准试验时表现更差;c)除正常老龄化以外的记忆减退的所有其它明显原因被排除(换句话说,记忆减退不能归因于其它原因,诸如近期心脏病发作或头部损伤,抑郁症,对药物的不良反应,阿尔茨海默病等)。
355、"年龄相关的认知减退(arcd)"表示作为人老龄化正常结果的记忆和认知能力减退(例如,craik&salthouse,1992)。这种情况实际上在所有哺乳动物种类中也是真实的。年龄相关的记忆损害表示相对于其年轻时代而言具有客观记忆减退,但认知功能相对于其同龄人而言是正常的老龄人(crook等人,1986)。年龄一致性记忆减退是强调这些是正常发育改变的较轻的恶化标记(crook,1993;larrabee,1996),是非病理生理性的(smith等人,1991)和罕有发展成明显的痴呆(youngjohn&crook,1993)。dsm-iv(1994)已经整理成arcd诊断分类。
356、"痴呆"表示这样一种病症,其特征在于干扰日常生活活动的严重认知缺陷。具有痴呆的受试者还展示出其它症状,诸如判断受损,人格改变,定向障碍,意识错乱,行为改变,语言困难和运动缺陷。存在不同类型的痴呆,诸如阿尔茨海默病(ad),血管性痴呆,痴呆伴露易小体(dementia with lewy bodies)和额颞叶痴呆。
357、阿尔茨海默病(ad)特征为在其早期阶段中的记忆缺陷。迟发症状包括受损的判断,定向障碍,意识错乱,行为变化,说话困难和运动缺陷。组织学上,ad特征为β-淀粉样斑块和蛋白tau的缠结。
358、血管性痴呆由中风引起。症状与ad的那些症状重叠,但是并没有多见于记忆损害。
359、痴呆伴露易小体特征为在脑中神经元内形成的α-突触核蛋白的异常沉着物。认知损害可能类似于ad,包括记忆和判断以及行为变化的损害。
360、额颞叶痴呆特征为在额皮层和/或前颞叶和皮克小体中神经胶质增生,神经元丧失,浅表海绵状变性。症状包括性格和行为的变化,其包括社会技能和语言表达/理解力的减退。
361、"创伤后应激障碍(ptsd)"表示特征为对突变事件立即或延迟反应的焦虑障碍,特征为再次经历创伤,心理麻痹或避免与创伤相关的刺激和增加的觉醒。再次经历现象包括对创伤提醒有侵入性记忆,幻觉重现,梦魇和心理或生理困扰的反应。这类反应产生焦虑,并可对患者的生活质量及身体和情感健康具有慢性和急性的显著影响。ptsd也与受损的认知表现相关,并且患ptsd的老龄个体相对于对照患者认知表现有更大的减退。
362、"精神分裂症"表示慢性虚弱障碍,特征为精神病理学谱,包括诸如异常或歪曲的心理特征(例如,幻觉,妄想)的阳性症状,特征为动机和适当的有意动作减少(例如,兴趣缺失,情感冷淡,无动机)和认知损害的阴性症状。尽管建议将脑的异常作为在精神分裂症中精神病理学的全谱基础,但目前可利用的抗精神病药大多对治疗患者认知损害无效。
363、"双相型障碍"或"bp"或"躁郁症"或"躁狂抑郁病"表示慢性心理/情感障碍,其可以表征为显著的情绪改变,包括抑郁期和陶醉性狂躁期。bp可以由本领域技术人员基于个人医疗史,访视会诊和身体检查来诊断。术语"躁狂"或"躁狂期"或其它变化形式表示其中个体显示如下特征的一些或全部的期限:快速思维,快速语言,活动水平升高和激动以及自我尊重感膨胀,陶醉感,判断力弱,失眠症,注意力不集中和进攻行为。
364、"肌萎缩性侧索硬化"也称为als,表示特征为运动神经元(在中枢神经系统中的神经细胞,其控制随意肌运动)变性的进行性,致死性,神经变性疾病。als也特征为在内嗅皮层和海马中神经元变性,记忆缺陷以及在诸如皮层的不同脑区中神经元兴奋性过高。
365、"与癌症治疗相关的认知损害"表示在用诸如化学治疗(例如化疗脑(chemobrain))和辐射的癌症治疗治疗的受试者中发展的认知损害。癌症治疗对脑的细胞毒性和其它不良副作用导致如记忆,学习和注意力的这类功能的认知损害。
366、帕金森病(pd)是特征在于随意运动减少的神经障碍。患病的患者与正常个体相比具有运动活动减少和较为缓慢的随意运动。该患者具有特征性"面具"脸,行走时的匆忙倾向,弯曲的体姿和肌肉泛发性虚弱。存在典型的"铅管样"强直的被动运动。该病的另一个重要特征在于在休息时的四肢震颤和运动过程中的减少。
367、本文中使用的"孤独症"表示孤独性谱群疾病,其特征在于受限和重复行为导致社会相互作用和沟通削弱的神经发育障碍。"孤独性谱群疾病"表示一组发育性残疾,其包括:孤独症;阿斯佩格综合征;未有特殊说明的全身性发育迟滞(pdd-nos或不典型孤独症);瑞特综合征;和童年瓦解性障碍。
368、精神发育迟缓是泛发性障碍,其特征在于显著受损的认知功能和适应行为缺陷。精神发育迟缓通常被定义为小于70的智商(iq)得分。在许多潜在的原因中,精神发育迟缓是先天性原因。神经元通讯功能障碍也被视为精神发育迟缓的潜在原因之一(myrrhe vanspronsen和casper c.hoogenraad,curr.neurol.neurosci.rep.2010,10,207-214)。
369、在某些情况下,精神发育迟缓包括但不限于唐氏综合征,腭-心-面综合征(velocardiofacial syndrome),胎儿酒精综合征,脆性x染色体综合征,克兰费尔特综合征,神经纤维瘤病,先天性甲状腺功能减退症,威廉斯综合征,苯丙酮尿(pku),史-伦-奥三氏综合征,帕-魏二氏综合征,phelan-mcdermid综合征,mowat-wilson综合征,纤毛类疾病(ciliopathy),lowe综合征和siderium型x-连锁智力障碍。唐氏综合征是一种障碍,其包括出生缺陷,包括一定程度的精神发育迟缓,特征性面部特征和通常的心脏缺陷,感染增加,与视觉和听觉相关的问题和其它健康问题的组合。脆性x染色体综合征是遗传性精神发育迟缓的普遍形式,其发生频率为4,000个男性中有1个和8,000个女性中有1个。这种综合征的特征还在于发展迟滞,活动过度,注意缺陷障碍和孤独样行为。对于脆性x染色体综合征没有有效的治疗方法。
370、强迫性障碍("ocd")是一种精神病症,其最常见的特征在于导致强迫行为的插入性的,反复性的不需要的思维(强迫观念)和个体感觉驱动行为(强迫)的心理活动。当前的流行病学数据表明ocd是美国第四位最常见的精神障碍。一些研究表明ocd的发病率在1%-3%之间,不过,临床公认的ocd的发病率低得多,这表明许多具有该障碍的个体可能未被诊断。具有ocd的患者通常由心理学家,精神病专家或精神分析学家根据《精神病诊断与统计手册》(diagnostic and statistical manual of mental disorders,第4版修订版(dsm-iv-tr)(2000)诊断标准(包括强迫观念行为和强迫的特征)来诊断。
371、物质成瘾(例如,药物成瘾,酒精成瘾)是一种精神障碍。这种成瘾并不在暴露物质滥用时被即时地触发。而是它牵涉随数小时至数日至数个月范围的不同时间过程发生的多个复杂的神经改变(kauer j.a.nat.rev.neurosci.2007,8,844-858)。成瘾的路径一般从自愿应用一种或多种受控的物质开始,诸如麻醉品,巴比妥酸盐类,甲基苯丙胺,酒精,烟碱和多种其它这样的管制物质中的任一种。随时间的推移,延长使用管制物质,自愿避开管制物质的能力因对脑功能和由此对行为的延长的影响而受损。这样,物质成瘾的特征一般在于强迫的物质渴求,寻找和使用,甚至在面对消极后果时仍然持续。渴求可以代表患者的潜在神经生物学改变,如果需要恢复,则必须能够以有意义的方式解决该患者的问题。在许多情况下,物质成瘾的特征还在于戒断症状,对于一些物质(例如,酒精,巴比妥酸盐类),所述戒断症状是威胁生命的,而在其它情况下,可能导致显著的病态(可以包括恶心,呕吐,发热,眩晕和大量出汗),痛苦和得到恢复的能力降低。例如,酒精中毒,也称作酒精依赖性是一种这样的物质成瘾。酒精中毒的特征主要在于4种症状,包括渴求,失控,身体依赖性和耐受性。这些症状还可以表征对其它管制物质成瘾。对酒精以及其它管制物质的渴求通常与对于食物或水的需求一样强烈。因此,尽管存在严重的家庭,健康和/或法律分歧,但是酒精可以持续饮用。
372、"治疗"病症或患者表示采取措施以得到有益的或期望的结果,包括临床结果。有益的或期望的临床结果包括但不限于预防或减慢疾病或障碍的发展,或减轻,改善或减慢与cns障碍相关的认知损害的一个或多个症状的发展,例如年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄相关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。在某些实施方案中,治疗包括预防或减慢cns障碍(例如本文所述的)的发展。在某些实施方案中,治疗包括减轻,改善或减慢与cns障碍相关的一个或多个症状的发展。在某些实施方案中,待治疗的症状是认知损害或认知缺陷。治疗年龄相关的认知损害还包括减慢年龄相关的认知损害(包括但不限于mci,arcd和aami)转化为痴呆(例如,ad)。
373、"治疗认知损害"表示采取措施以改善在患认知损害的受试者中的认知功能,以使一个或多个认知试验中受试者的表现改善至任何可检测的程度,或预防进一步减退。优选地,在治疗认知损害之后,受试者的认知功能更接近类似于正常,未受损的受试者的功能。治疗在人类中的认知损害可将认知功能改善至任何可检测的程度,但是优选改善至足以允许受损受试者以与正常未受损的受试者相同的熟练水平进行每日正常生活的活动。在一些情况下,"治疗认知损害"表示采取措施以改善在患认知损害的受试者中的认知功能,以使一个或多个认知试验中受试者的表现改善至任何可检测的程度,或预防进一步减退。优选地,在治疗认知损害之后,受试者的认知功能更接近类似于正常,未受损的受试者的功能。在一些情况下,在受到年龄相关的认知损害影响的受试者中"治疗认知损害"表示采取措施以改善该受试者中的认知功能,以使在治疗认知损害之后受试者的认知功能更接近类似于年龄匹配的正常,未受损受试者的功能,或年轻成年受试者的功能。
374、物质,化合物或试剂对受试者的"施用"((administering)或(administration))可使用本领域技术人员已知的多种方法之一进行。例如,可通过静脉内,动脉,皮内,肌内,腹膜内,静脉内,皮下,眼部,舌下,口服(通过摄入),鼻内(通过吸入),脊柱内,脑内和透皮(通过吸收,例如通过皮肤输送管)施用化合物或试剂。还可通过可再加入(rechargeable)或可生物降解的聚合物装置或其它装置诸如贴剂和泵或制剂适当导入化合物或试剂,这提供延长,减慢或受控的化合物或试剂释放。施用还可进行诸如一次,多次和/或在一个或多个延长期限内进行。在一些方面,施用包括直接施用,包括自身施用;和间接施用,包括为药物开处方行为。例如,作为本文使用的,指导患者自身施用或由另一个人施用药物和/或给患者提供药物处方的临床医师对患者施用药物。
375、物质,化合物或试剂对受试者的适合的施用方法还取决于例如受试者年龄,无论该受试者在施用时是活动的还是不活动的,无论受试者在施用时是否是认识受损的;损害程度和化合物或试剂的化学和生物特性(例如溶解性,可消化性,生物利用度,稳定性和毒性)。在某些实施方案中,通过口服(例如通过摄入)或静脉内(例如,通过注射)对受试者施用化合物或试剂。在某些实施方案中,口服施用的化合物或试剂是延长释放(extendedrelease)或缓慢释放制剂的形式或使用用于这种缓慢释放或延长释放的装置施用。
376、本文中使用的"含有α5的gabaa受体激动剂","含有α5的gabaa r激动剂"或"gabaaα5受体激动剂"和本文使用的其它变型表示增强含有α5的gabaa受体(gabaa r)的功能的化合物,即增加gaba-门控cl-电流的化合物。在某些实施方案中,本文使用的含有α5的gabaa r激动剂表示增强gaba活性的正变构调节剂。适合用在本发明中的含有α5的gabaa受体激动剂包括所有通式的含有α5的gabaa受体激动剂和本文所述的特定的含有α5的gabaa受体激动剂及它们的水合物,溶剂化物,多晶型物,盐(例如,药学上可接受的盐),异构体(例如,立体异构体,e/z异构体和互变异构体)及其组合。
377、"抗精神病的","抗精神病剂","抗精神病药"或"抗精神病化合物"表示(1)典型的或非典型抗精神病药物;(2)试剂,其选自多巴胺能药,谷氨酸能药,nmda受体正变构调节剂,甘氨酸再摄取抑制剂,谷氨酸再摄取抑制剂,亲代谢性谷氨酸盐受体(mglurs)激动剂或正变构调节剂(pams)(例如,mglur2/3激动剂或pams),谷氨酸受体glur5正变构调节剂(pams),m1蕈毒碱乙酰胆碱受体(machr)正变构调节剂(pams),组胺h3受体拮抗剂,ampa/红藻氨酸盐受体拮抗剂,安帕金(cx-516),谷胱甘肽前药,去甲肾上腺素能药,血清素受体调节剂,胆碱能药,大麻素cb1拮抗剂,神经激肽3拮抗剂,神经降压肽激动剂,mao b抑制剂,pde10抑制剂,nnos抑制剂,神经递质药和神经营养因子,α-7激动剂或正变构调节剂(pams)pams,血清素2c激动剂;和/或(3)可用于治疗精神分裂症或双相型障碍(特别是躁狂症)的一种或多种征象或症状的试剂。
378、本文中使用的"典型的抗精神病药"表示常规的抗精神病药,其产生抗精神病作用以及与黑质纹状体多巴胺系统的紊乱有关的活动相关副作用。这些锥体束外的副作用(eps)包括帕金森综合征,静坐不能,迟发性运动障碍和张力障碍。参见baldessarini和tarazi,goodman&gilman'sthe pharmacological basis of therapeutics 10edition,2001,第485-520页。
379、本文中使用的"非典型抗精神病药"表示具有少量eps或无eps的产生抗精神病作用的抗精神病药,其包括但不限于阿立哌唑,阿塞那平,氯氮平,伊潘立酮,奥氮平,鲁拉西酮,帕潘立酮,喹噻平,维思通和齐拉西酮。"非典型"抗精神病药的药理学特性不同于常规抗精神病药。虽然常规抗精神病药的特征主要是d2多巴胺受体阻断,但是非典型抗精神病药显示对多个受体的拮抗作用,所述受体包括5hta和5htc血清素受体受体并且受体亲和力的程度不同。非典型抗精神病药通常表示血清素/多巴胺拮抗剂,其反映了一个有影响力的假说,即,与d2受体相比对5ht2受体更高的亲和力是"非典型"抗精神病药物作用或"第二代"抗精神病药物的基础。但是,非典型抗精神病药通常显示副作用,其包括但不限于体重增加,糖尿病(例如,ii型糖尿病),高脂血症,qtc间期延长,心肌炎,性功能副作用,锥体束外的副作用和白内障。所以,考虑到非典型抗精神病药在临床症状的减轻和它们导致诸如以上所列那些副作用的可能性方面的不同,非典型抗精神病药不代表一个相同的类型。另外,如上文所述的非典型抗精神病药的通常的副作用通常限制可用于这些药剂的所述抗精神病药的剂量。
380、美金刚的化学名称是3,5-二甲基金刚烷-1-胺或3,5-二甲基三环[3.3.1.13,7]癸烷-1-胺,其是非竞争性n-甲基-d-天冬氨酸(nmda)受体拮抗剂,具有中等的亲和力。美金刚的专有名称包括:和(merz),(forestlaboratories),和(lundbeck)和(unipharm)。美金刚在美国被批准用于治疗中度至重度阿尔茨海默病(ad),剂量为至多28mg/天。美金刚的衍生物或类似物包括结构或化学上类似于美金刚的化合物,它们也可用于本发明。所述美金刚的衍生物或类似物包括但不限于美国专利号3,391,142,4,122,193,4,273,774和5,061,703,美国专利申请公开us20040087658,us20050113458,us20060205822,us20090081259,us20090124659和us20100227852,欧洲专利申请公开ep2260839a2,欧洲专利ep1682109b1和pct申请公开wo2005079779中所述的那些化合物,所有这些文献都通过引用并入本文。本发明中所用的美金刚包括美金刚及其衍生物和类似物,以及其水合物,多晶型物,前药,盐和溶剂化物。本文所用的美金刚还包括包含美金刚或衍生物或类似物或其药学上可接受的盐,水合物,溶剂化物,多晶型物或前药的组合物,其中所述组合物任选地还包含至少一种另外的治疗剂(诸如可用于治疗cns障碍或其相关认知损害的治疗剂)。在某些实施方案中,适合用在本发明中的所述美金刚组合物包含美金刚和第二种治疗剂,所述第二种治疗剂是多奈哌齐(商品名aricept)。
381、本文中使用的"乙酰胆碱酯酶抑制剂"或"ache-i"表示抑制乙酰胆碱酯酶分解神经递质乙酰胆碱的能力的试剂,由此增加主要在脑突触或神经肌肉接头的乙酰胆碱的浓度或持续时间。适合用于本技术的ache-i可包括,例如(i)可逆的非竞争性抑制剂或可逆的竞争性抑制剂,(ii)不可逆的和(iii)准不可逆的抑制剂的亚类。
382、本文中使用的术语"同时施用"是指,将含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)和第二种治疗剂(例如,抗精神病药,美金刚或ache-i)或它们的药学上可接受的盐,水合物,溶剂化物或多晶型物以不多于约15分钟,且在某些实施方案中不多于约10分钟的时间间隔施用。当同时施用药物时,可将含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)和第二种治疗剂(例如,抗精神病药,美金刚或ache-i)或它们的盐,水合物,溶剂化物或多晶型物包含在相同剂量中(例如,包含含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)和第二种治疗剂(例如,抗精神病药,美金刚或ache-i)的单元剂型),或者包含在离散的剂量中(例如,含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)或其盐,水合物,溶剂化物或多晶型物包含在一个剂型中,且第二种治疗剂(例如,抗精神病药,美金刚或ache-i)或其盐,水合物,溶剂化物或多晶型物包含在另一个剂型中)。
383、本文中使用的术语"依次施用"表示将含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)和第二种治疗剂(例如,抗精神病药,美金刚或ache-i)或它们的药学上可接受的盐,水合物,溶剂化物,多晶型物以多于约15分钟,且在某些实施方案中多于约1小时,或至多12-24小时的时间间隔施用。可首先施用含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂),或者首先施用第二种治疗剂(例如,抗精神病药,美金刚或ache-i)。对于依次施用,含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂)和第二种治疗剂(例如,抗精神病药,美金刚或ache-i)或它们的盐,水合物,溶剂或多晶型物可以包含在离散的剂型中,任选地包含在相同的容器或包装中。
384、药物或试剂的"治疗有效量"是在对受试者施用时具有指定治疗效果(例如改善受试者认知功能,所述受试者例如为患有与cns障碍相关的认知损害的患者)的药物或试剂用量。完全的治疗效果不一定通过施用一次剂量出现且可能在施用一系列剂量之后出现。因此,可以以一次或多次施用来施用治疗有效量。受试者所需的精确有效量将取决于例如受试者体型,健康状况和年龄,认知损害或cns障碍的其它症状(例如年龄相关的认知损害,轻度认知损害(mci),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相障碍(bipolar),als,与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾)的性质和程度,以及为施用和施用方式选择的治疗剂或治疗剂组合。本领域技术人员易于通过常规实验确定用于指定情况的有效量。
385、本发明的化合物还包括前药,类似物或衍生物。术语"前药"是公认的和意欲包括在生理条件下被转化成含有α5的gabaa r正变构调节剂的化合物或试剂。制备前药的常用方法在于选择在生理条件下水解或代谢的部分,以提供期望化合物或试剂。在其它实施方案中,前药通过宿主动物的酶活性被转化成gabaaα5正变构调节剂。
386、"类似物"在本文中用于表示功能上类似于另一个化学实体,但是并没有共有相同化学结构的化合物。例如,类似物十分类似于基础或母体化合物,以至于尽管结构上稍有不同,它也可在治疗应用中替代基础化合物。
387、"衍生物"在本文中用于表示化合物的化学修饰。化合物的化学修饰可包括,例如,用烷基,酰基或氨基替代氢。许多其它修饰也是可能的。
388、本文中使用的术语"脂族基"表示直链或支链烷基,烯基或炔基。应理解,烯基或炔基实施方案要求在脂族基链上至少有2个碳原子。脂族基典型地包含1(或2)-12个碳,诸如1(或2)-4个碳。
389、本文中使用的术语"芳基"表示单环或双环的碳环芳环系统。本文中使用的芳基包括(c6-c12)-芳基-。例如,本文中使用的芳基可以是c6-c10单环或c8-c12二环的碳环芳环系统。在某些实施方案中,本文中使用的芳基可以是(c6-c10)-芳基-。苯基(或ph)是单环芳环系统的例子。二环芳环系统包括这样的系统,其中2个环都是芳族的,例如萘基,和这样的系统,其中2个环中仅1个是芳族的,例如四氢萘。
390、本文中使用的术语"杂环"表示以化学稳定的排列方式具有1-4个选自o,n,nh,s,so或so2的杂原子或杂原子基团的单环或二环非芳环系统。本文中使用的杂环包括3-12元的杂环基,其具有1-4个独立地选自o,n,nh,s,so或so2的杂原子。例如,本文中使用的杂环可以是3-10元的单环或8-12元的二环的非芳环系统,其以化学稳定的排列方式在各个环中具有1-4个选自o,n,nh,s,so或so2的杂原子或杂原子基团。在某些实施方案中,本文中使用的杂环可以是3-10元的杂环基-,其具有1-4个独立地选自o,n,nh,s,so或so2的杂原子。在"杂环基"的二环非芳环系统的实施方案中,一个或两个环可以包含所述杂原子或杂原子基团。在另一个二环"杂环基"的实施方案中,2个环中的1个可以是芳族的。在又另一个杂环环系的实施方案中,非芳族杂环可以任选地与芳族碳环稠合。
391、杂环的例子包括3-1h-苯并咪唑-2-酮,3-(1-烷基)-苯并咪唑-2-酮,2-四氢呋喃基,3-四氢呋喃基,2-四氢噻吩基,3-四氢噻吩基,2-吗啉代,3-吗啉代,4-吗啉代,2-硫代吗啉代,3-硫代吗啉代,4-硫代吗啉代,1-吡咯烷基,2-吡咯烷基,3-吡咯烷基,1-四氢哌嗪基,2-四氢哌嗪基,3-四氢哌嗪基,1-哌啶基,2-哌啶基,3-哌啶基,1-吡唑啉基,3-吡唑啉基,4-吡唑啉基,5-吡唑啉基,1-哌啶基,2-哌啶基,3-哌啶基,4-哌啶基,2-噻唑烷基,3-噻唑烷基,4-噻唑烷基,1-咪唑烷基,2-咪唑烷基,4-咪唑烷基,5-咪唑烷基,吲哚啉基,四氢喹啉基,四氢异喹啉基,苯并硫杂环戊烷(benzothiolane),苯并二噻烷(benzodithiane)和1,3-二氢-咪唑-2-酮。
392、本文中使用的术语"杂芳基"表示以化学稳定的排列方式具有选自o,n,nh或s的1-4个杂原子或杂原子基团的单环或二环芳环系统。本文中使用的杂芳基包括5-12元的杂芳基,其具有1-4个独立地选自o,n,nh或s的杂原子。在某些实施方案中,本文中使用的杂芳基可以是5-10元的杂芳基,其具有1-4个独立地选自o,n,nh或s的杂原子。例如,本文中使用的杂芳基可以是5-10元的单环或8-12元的二环的芳环系统,其以化学稳定的排列方式在一个或两个环中具有1-4个选自o,n,nh或s的杂原子或杂原子基团。在"杂芳基"的这样的一个二环芳环系统实施方案中:
393、-两个环都是芳族的;且
394、-一个或两个环可以含有所述杂原子或杂原子基团。
395、杂芳基环的例子包括2-呋喃基,3-呋喃基,n-咪唑基,2-咪唑基,4-咪唑基,5-咪唑基,苯并咪唑基,3-异噁唑基,4-异噁唑基,5-异噁唑基,2-噁唑基,4-噁唑基,5-噁唑基,n-吡咯基,2-吡咯基,3-吡咯基,2-吡啶基,3-吡啶基,4-吡啶基,2-嘧啶基,4-嘧啶基,5-嘧啶基,哒嗪基(例如,3-哒嗪基),2-噻唑基,4-噻唑基,5-噻唑基,四唑基(例如,5-四唑基),三唑基(例如,2-三唑基和5-三唑基),2-噻吩基,3-噻吩基,苯并呋喃基,苯并噻吩基,吲哚基(例如,2-吲哚基),吡唑基(例如,2-吡唑基),异噻唑基,1,2,3-噁二唑基,1,2,5-噁二唑基,1,2,4-噁二唑基,1,2,3-三唑基,1,2,3-噻二唑基,1,3,4-噻二唑基,1,2,5-噻二唑基,嘌呤基,吡嗪基,1,3,5-三嗪基,喹啉基(例如,2-喹啉基,3-喹啉基,4-喹啉基)和异喹啉基(例如,1-异喹啉基,3-异喹啉基或4-异喹啉基)。
396、术语"环烷基或环烯基"表示单环或稠合或桥连双环碳环环系,其为非芳族的。例如,本文所用的环烷基或环烯基可以是c3-c10单环或稠合或桥连c8-c12双环碳环环系,其不是芳族的。环烯基环可以具有一个或多个不饱和度。优选的环烷基或环烯基包括环丙基,环丁基,环戊基,环己基,环己烯基,环庚基,环庚烯基,降冰片基,金刚烷基和十氢萘基。
397、术语"杂芳烷基"表示烷基,其中杂芳基取代烷基h原子。例如,[[[杂芳基=杂环和芳族]]]
398、本文所用的碳原子命名可以具有所示的整数和任意插入的整数。例如,(c1-c4)-烷基上的碳原子数是1,2,3或4。应理解,这些命名表示适合基团上的总原子数。例如,在(c3-c10)-杂环基上,碳原子和杂原子总数是3(如在氮丙啶中),4,5,6(如在吗啉中),7,8,9或10。
399、"药学上可接受的盐"在本文中用于表示根据本发明的试剂或化合物,其为化合物的治疗活性的无毒性碱式盐和酸式盐的形式。作为游离碱形式存在的化合物的酸加成盐形式可以通过用适合的酸处理所述游离碱形式得到,所述适合的酸诸如是无机酸,例如氢卤酸,诸如氢氯酸或氢溴酸,硫酸,硝酸,磷酸等;或有机酸,例如乙酸,羟基乙酸,丙酸,乳酸,丙酮酸,丙二酸,丁二酸,马来酸,富马酸,苹果酸,酒石酸,柠檬酸,甲磺酸,乙磺酸,苯磺酸,对甲苯磺酸,环状酸(cyclic),水杨酸,对氨基水杨酸,双羟萘酸等。参见,例如wo 01/062726。
400、可以通过用适合的有机碱和无机碱处理将包含酸性质子的化合物转化成其治疗活性的无毒性的碱加成盐形式,例如金属盐或胺盐。适合的碱式盐形式包括,例如铵盐,碱金属和碱土金属盐例如锂,钠,钾,镁,钙盐等,与有机碱的盐,例如n-甲基-d-葡萄糖胺,海巴明盐和与氨基酸(例如精氨酸,赖氨酸等)的盐。相反,可以通过用适合的碱或酸处理将所述盐形式转化成游离形式。
401、化合物及其盐可以是溶剂化物形式,其包括在本发明范围内。这种溶剂化物包括,例如水合物,醇化物等。参见,例如wo 01/062726。
402、本文中使用的术语"水合物"表示水与化合物的组合,其中所述水保持与水一样的分子状态,并且是吸收的,吸附的或包含在该基底化合物的晶格中。
403、本文中使用的术语"多晶型物"表示相同化合物的不同晶形和其它固态分子形式,其包括相同化合物的假-多晶型物,诸如水合物(例如,在晶体结构中存在的结合水)和溶剂化物(例如,非水的结合溶剂)。由于分子在晶格中的不同堆积,不同的晶状的多晶型物具有不同的晶体结构。这导致直接影响晶体的物理性质如晶体或粉末的x射线衍射特性的不同的晶体对称性和/或晶胞参数。例如,不同的多晶型物通常会以不同的一组角度衍射,并产生不同的强度值。因此,x射线粉末衍射可用于以可再现且可靠的方式鉴定不同多晶型物或包含多于一种多晶型物的固体形式。结晶性多晶型形式是为制药产业特别是对于参与开发合适的剂型的制药产业所关注的。如果多晶型形式在临床研究或稳定性研究中并未保持恒定,则所用或所研究的确切剂型在批次之间不具有可比性。当将化合物用于临床研究或商业产品时,还期望产生具有高纯度的所选多晶型形式的化合物的方法,因为存在的杂质可能产生不期望的毒性作用。某些多晶型形式可能表现出增强的热力学稳定性或可能更易于以高纯度大量制造,并且因此更适于包含在药物制剂中。由于不同的晶格能量,某些多晶型物可能表现出其它有利的物理性质,如不具有吸湿倾向,溶解性改善以及溶出速率提高。
404、本技术预见到式i-iv的化合物的所有异构体。本文中使用的"异构体"包括光学异构体(例如立体异构体,例如,对映异构体和非对映异构体),z(zusammen)或e(entgegen)异构体和互变异构体。用于本发明的方法和组合物的许多化合物在其结构上具有至少一个立体中心。该立体中心可以以r或s构型存在,所述r和s标识与pure appl.chem.(1976),45,11-30中所述的规则相符。本发明还涉及所有立体异构体形式,诸如化合物的对映异构体和非对映异构体形式或其混合物(包括所有可能的立体异构体混合物)。参见,例如,wo 01/062726。此外,包含烯基的一些化合物可以作为z(zusammen)或e(entgegen)异构体存在。在每种情况下,本发明包括混合物和单独的各异构体。哌啶基或氮杂庚环基环上的多个取代基还可以表示彼此在哌啶基或氮杂庚环基环平面上的顺式或反式相关性。一些化合物还可以以互变体形式存在。尽管本文所述的通式中未明确表示,但是这种形式意欲包括在本发明范围内。就本发明的方法和组合物而言,所涉及的化合物意欲包括每种其可能的异构体形式的化合物及其混合物,除非特别指定具体的异构体形式。参见,例如,wo01/062726。
405、本发明的化合物增强含有α5的gabaa r的功能,即,它们是含有α5的gabaa r激动剂(例如,含有α5的gabaa受体正变构调节剂),并能增加gaba-门控cl-电流。
406、本发明还提供了药物组合物,其包含一种或多种本发明的化合物以及药学上可接受的载体或赋形剂。在某些实施方案中,本技术的药物组合物还可以包含第二种治疗剂,例如抗精神病药,美金刚或ache-i。
407、本发明还提供了治疗与所述cns障碍有关的认知损害的方法,所述cns障碍对含有α5的gabaa受体的正变构调节剂有响应,例如,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。在某些实施方案中,所述方法是治疗年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾的方法。在某些实施方案中,治疗包括预防或减慢如本文所述的cns障碍(诸如本文描述的那些)的发展。在某些实施方案中,治疗包括减轻,改善或减慢与cns障碍有关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害或认知缺陷。在本发明的另一个方面,提供了在有此需要的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
408、具有认知损害的各种cns障碍(例如,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾)可具有多种病因。然而,在每种上述障碍中的认知损害症状可能具有重叠的原因。因此,治疗一种cns障碍中的认知损害的组合物或治疗方法还可以治疗另一种情况下的认知损害。
409、苯并二氮杂环庚三烯衍生物
410、本发明提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
411、
412、其中:
413、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
414、a是c,cr6或n;
415、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
416、d是n,nr7,o,cr6或c(r6)2;
417、e是n,nr7,cr6或c(r6)2;
418、w是n,nr7,cr6或c(r6)2;
419、x是n,nr7,o,cr6或c(r6)2;
420、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
421、v是c或cr6,
422、或者当z是c或cr6时,v是c,cr6或n;
423、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10;且其中r2独立地被0-5个r’取代;
424、m和n独立地是选自0-4的整数;
425、p是选自2-4的整数;
426、键的每次出现是单键或双键;
427、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
428、r3不存在或选自:
429、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,
430、
431、-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
432、每个r6独立地是-h或-(c1-c6)烷基;
433、每个r7独立地是-h或-(c1-c6)烷基;
434、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
435、每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;
436、每个r独立地选自:
437、h-,
438、(c1-c12)-脂族基-,
439、(c3-c10)-环烷基-,
440、(c3-c10)-环烯基-,
441、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
442、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
443、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
444、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
445、(c6-c10)-芳基-,
446、(c6-c10)-芳基-(c1-c12)脂族基-,
447、(c6-c10)-芳基-o-(c1-c12)脂族基-,
448、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
449、3-10元杂环基-,
450、(3-10元杂环基)-(c1-c12)脂族基-,
451、(3-10元杂环基)-o-(c1-c12)脂族基-,
452、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
453、5-10元杂芳基-,
454、(5-10元杂芳基)-(c1-c12)-脂族基-,
455、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
456、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
457、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
458、其中r的每次出现独立地被0-5个r’取代;
459、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
460、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
461、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2nro 2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
462、在某些实施方案中,本发明提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
463、
464、其中:
465、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
466、a是c,cr6或n;
467、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
468、d是n,nr7,o,cr6或c(r6)2;
469、e是n,nr7,cr6或c(r6)2;
470、w是n,nr7,cr6或c(r6)2;
471、x是n,nr7,o,cr6或c(r6)2;
472、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
473、v是c或cr6,
474、或者当z是c或cr6时,v是c,cr6或n;
475、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10;且其中r2独立地被0-5个r’取代;
476、m和n独立地是选自0-4的整数;
477、p是选自2-4的整数;
478、键的每次出现是单键或双键;
479、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
480、r3不存在或选自:
481、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
482、每个r6独立地是-h或-(c1-c6)烷基;
483、每个r7独立地是-h或-(c1-c6)烷基;
484、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
485、每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;
486、每个r独立地选自:
487、h-,
488、(c1-c12)-脂族基-,
489、(c3-c10)-环烷基-,
490、(c3-c10)-环烯基-,
491、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
492、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
493、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
494、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
495、(c6-c10)-芳基-,
496、(c6-c10)-芳基-(c1-c12)脂族基-,
497、(c6-c10)-芳基-o-(c1-c12)脂族基-,
498、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
499、3-10元杂环基-,
500、(3-10元杂环基)-(c1-c12)脂族基-,
501、(3-10元杂环基)-o-(c1-c12)脂族基-,
502、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
503、5-10元杂芳基-,
504、(5-10元杂芳基)-(c1-c12)-脂族基-,
505、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
506、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
507、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
508、其中r的每次出现独立地被0-5个r’取代;
509、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
510、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
511、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
512、某些实施方案提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
513、
514、其中:
515、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
516、a是c,cr6或n;
517、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
518、d是n,nr7,o,cr6或c(r6)2;
519、e是n,nr7,cr6或c(r6)2;
520、w是n,nr7,cr6或c(r6)2;
521、x是n,nr7,o,cr6或c(r6)2;
522、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
523、v是c或cr6,
524、或者当z是c或cr6时,v是c,cr6或n;
525、其中当由x,y,z,v和w形成的环是时,那么r2是-or8,-sr8或-(ch2)nor8;
526、m和n各自独立地是选自0-4的整数;
527、键的每次出现是单键或双键;
528、r1,r2,r4和r5的每次出现各自独立地选自:卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
529、r3不存在或选自:
530、卤素,-r,-or,-no2,-ncs,-cn,-cf3,-ocf3,-sir3,-n(r)2,-sr,-sor,-so2r,-so2n(r)2,-so3r,-(cr2)1-3r,-(cr2)1-3-or,-(cr2)0-3-c(o)nr(cr2)0-3r,-(cr2)0-3-c(o)nr(cr2)0-3or,-c(o)r,-c(o)c(o)r,-c(o)ch2c(o)r,-c(s)r,-c(s)or,-c(o)or,-c(o)c(o)or,-c(o)c(o)n(r)2,-oc(o)r,-c(o)n(r)2,-oc(o)n(r)2,-c(s)n(r)2,-(cr2)0-3nhc(o)r,-n(r)n(r)cor,-n(r)n(r)c(o)or,-n(r)n(r)con(r)2,-n(r)so2r,-n(r)so2n(r)2,-n(r)c(o)or,-n(r)c(o)r,-n(r)c(s)r,-n(r)c(o)n(r)2,-n(r)c(s)n(r)2,-n(cor)cor,-n(or)r,-c(=nh)n(r)2,-c(o)n(or)r,-c(=nor)r,-op(o)(or)2,-p(o)(r)2,-p(o)(or)2和-p(o)(h)(or);
531、每个r6独立地是-h或-(c1-c6)烷基;
532、每个r7独立地是-h或-(c1-c6)烷基;
533、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
534、每个r独立地选自:
535、h-,
536、(c1-c12)-脂族基-,
537、(c3-c10)-环烷基-,
538、(c3-c10)-环烯基-,
539、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
540、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
541、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
542、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
543、(c6-c10)-芳基-,
544、(c6-c10)-芳基-(c1-c12)脂族基-,
545、(c6-c10)-芳基-o-(c1-c12)脂族基-,
546、3-10元杂环基-,
547、(3-10元杂环基)-(c1-c12)脂族基-,
548、(3-10元杂环基)-o-(c1-c12)脂族基-,
549、5-10元杂芳基-,
550、(5-10元杂芳基)-(c1-c12)-脂族基-和
551、(5-10元杂芳基)-o-(c1-c12)-脂族基-;
552、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
553、其中r的每次出现独立地被0-5个r’取代;
554、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
555、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
556、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
557、本发明提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
558、
559、其中:
560、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
561、a是c,cr6或n;
562、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
563、d是n,nr7,o,cr6或c(r6)2;
564、e是n,nr7,cr6或c(r6)2;
565、w是n,nr7,cr6或c(r6)2;
566、x是n,nr7,o,cr6或c(r6)2;
567、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
568、v是c或cr6,
569、或者当z是c或cr6时,v是c,cr6或n;
570、其中当由x,y,z,v和w形成的环是时,那么r2是-(ch2)nor8或-(ch2)no(ch2)nr8;且其中r2独立地被0-5个r’取代;
571、m和n独立地是选自0-4的整数;
572、p是选自2-4的整数;
573、键的每次出现是单键或双键;
574、每个r1独立地选自:卤素,-r和-or;
575、r2选自:卤素,-r和-(cr2)1-3-or;
576、r3选自:-r和-cn;
577、r4和r5各自独立地是-h或-(c1-c6)烷基;
578、每个r6独立地是-h或-(c1-c6)烷基;
579、每个r7独立地是-h或-(c1-c6)烷基;
580、每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;
581、每个r独立地选自:
582、h-,
583、(c1-c12)-脂族基-,
584、(c3-c10)-环烷基-,
585、(c3-c10)-环烯基-,
586、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
587、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
588、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
589、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
590、(c6-c10)-芳基-,
591、(c6-c10)-芳基-(c1-c12)脂族基-,
592、(c6-c10)-芳基-o-(c1-c12)脂族基-,
593、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
594、3-10元杂环基-,
595、(3-10元杂环基)-(c1-c12)脂族基-,
596、(3-10元杂环基)-o-(c1-c12)脂族基-,
597、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
598、5-10元杂芳基-,
599、(5-10元杂芳基)-(c1-c12)-脂族基-,
600、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
601、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
602、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
603、其中r的每次出现独立地被0-5个r’取代;
604、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
605、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
606、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
607、本发明提供了式i的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
608、
609、其中:
610、u和由α和β标示的两个碳原子一起形成具有0-2个氮原子的5或6元芳族环;
611、a是c,cr6或n;
612、b和f各自独立地选自c,cr6和n,其中b和f不可都是n;
613、d是n,nr7,o,cr6或c(r6)2;
614、e是n,nr7,cr6或c(r6)2;
615、w是n,nr7,cr6或c(r6)2;
616、x是n,nr7,o,cr6或c(r6)2;
617、y和z各自独立地选自c,cr6和n,其中y和z不可都是n;
618、v是c或cr6,
619、或者当z是c或cr6时,v是c,cr6或n;
620、其中当由x,y,z,v和w形成的环是时,那么r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是-(c1-c6)烷基或(c6-c10)-芳基(例如苯基),且其中r2独立地被0-5个r’取代;
621、m和n独立地是选自0-4的整数(在某些实施方案中,m是1);
622、p是选自2-4的整数;
623、键的每次出现是单键或双键;
624、每个r1独立地选自:-cl,-f,-ome和-c≡ch;
625、r2是卤素,-(cr2)1-3-or,其中r的每次出现独立地选自-h,-(c1-c6)烷基,(c6-c10)-芳基-(例如苯基)和(c6-c10)-芳基-(c1-c12)脂族基-(例如苯基-(c1-c6)烷基-),且其中r的每次出现独立地被0-5个r’取代;
626、r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-c≡c-苯基,
627、其中r3被0-5个r’取代;
628、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
629、每个r6独立地是-h或-(c1-c6)烷基;
630、每个r7独立地是-h或-(c1-c6)烷基;
631、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
632、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2nro 2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
633、在以上实施方案中的某些中,r3选自:
634、
635、其中r”的每次出现独立地选自-(c1-c6)-烷基(例如,直链或支链),-c≡ch,苯基,噻吩,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,其中每个r”独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2nro 2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
636、在式i的化合物的某些实施方案中,x,y,z,v和w一起形成具有1-4个氮原子的5元芳族或非芳族环,其中所述环被0-3个r6和0-2个r7取代。在某些实施方案中,x,y,z,v和w一起形成具有1-3个氮原子的5元芳族环,其中所述环被0-2个r6和0-1个r7取代。
637、在某些实施方案中,x,y,z,v和w形成选自以下的环:
638、
639、在某些实施方案中,x,y,z,v和w形成选自以下的环:
640、
641、在式i的化合物的某些实施方案中,w是n。在某些实施方案中,w是n,且x,y,z,v和w形成选自以下的环:
642、
643、在某些实施方案中,w是n,且x,y,z,v和w形成选自以下的环:
644、
645、在式i的化合物的某些实施方案中,由x,y,z,v和w形成的环是:
646、
647、在式i的化合物的某些实施方案中,由x,y,z,v和w形成的环是:
648、
649、在式i的化合物的某些实施方案中,由x,y,z,v和w形成的环选自:
650、
651、在式i的化合物的某些实施方案中,由x,y,z,v和w形成的环选自:
652、
653、在某些实施方案中,由x,y,z,v和w形成的环是:在某些实施方案中,由x,y,z,v和w形成的环是:
654、
655、在式i的化合物的某些实施方案中,a,b,d,e和f一起形成具有1-4个氮原子的5元芳族或非芳族环,其中所述环被0-3个r6和0-2个r7取代。在某些实施方案中,a,b,d,e和f一起形成具有1-3个氮原子的5元芳族环,其中所述环被0-2个r6和0-1个r7取代。
656、在式i的化合物的某些实施方案中,a,b,d,e和f形成选自以下的环:
657、
658、在式i的化合物的某些实施方案中,由a,b,d,f和e形成的环是:
659、
660、在式i的化合物的某些实施方案中,所述化合物具有式ii的结构:
661、
662、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中m,r1,r2,r3,r4,r5和r6如在式i中所定义。
663、在式i的化合物的某些实施方案中,所述化合物具有式iii的结构:
664、
665、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中m,r1,r2,r3,r4,r5和r6如在式i中所定义。
666、在式i的化合物的某些实施方案中,所述化合物具有式iv的结构:
667、
668、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中r2是-or8,-sr8或-(ch2)nor8,其中r2独立地被0-5个r’取代,且其中m,n,r1,r3,r4,r5,r6和r8如在式i中所定义。在某些实施方案中,r2是-or8。在某些实施方案中,r2是-(ch2)nor8。
669、在式i的化合物的某些实施方案中,所述化合物具有式iv的结构:
670、
671、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中r2是-(ch2)no(ch2)nr8,-(ch2)pr8或-(ch2)nn(r”)r10,其中r2独立地被0-5个r’取代,且其中m,n,p,r1,r3,r4,r5,r6,r8,r10和r”如本文中定义。在某些实施方案中,r2是-(ch2)no(ch2)nr8。
672、在式i,ii,iii或iv的化合物的某些实施方案中,r1的每次出现选自:卤素,-r,-or,-no2,-cn,-cf3,-ocf3,-n(r)2和-n(r)so2r,其中r的每次出现独立地被0-5个r’取代。在某些实施方案中,r1的每次出现独立地选自:卤素,-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-no2,-cn,-cf3,-ocf3,-nh2,-n((c1-c6)烷基)2,-n((c1-c6)烷基)so2((c1-c6)烷基)和-nhso2((c1-c6)烷基),其中所述烷基独立地被0-5个r’取代。在某些实施方案中,r1的每次出现独立地选自:-h,-f,-cl,-br,-oh,-me,-et,-ome,-oet,-no2,-cn,-cf3,-ocf3,-nh2,-nme2,-net2,-nhso2me和-nhso2et。在式i-iv中的任一个的化合物的某些实施方案中,至少一个r1是-or。在某些实施方案中,至少一个r1是-o((c1-c6)烷基),诸如-ome。
673、在式i,ii或iii的化合物的某些实施方案中,r2选自:卤素,-r,-or,-no2,-(cr2)1-3r,-(cr2)1-3-or,-cn,-cf3,-c(o)nr2,-c(o)or和-ocf3,其中r的每次出现独立地被0-5个r’取代。在某些实施方案中,r2选自:-h,-(c1-c6)烷基,-ch2-o((c1-c6)烷基),-(c((c1-c6)烷基)2)1-3-o((c1-c6)烷基),-oh,-o((c1-c6)烷基),-no2,-cn,-cf3,-ocf3,(c3-c10)-环烷基-,-c(o)n((c1-c6)烷基)2,-c(o)o((c1-c6)烷基),3-10元杂环基-,(c6-c10)芳基-,5-10元杂芳基-,(c6-c10)芳基-(c1-c12)脂族基-,(c6-c10)芳基-o-(c1-c12)脂族基-,(c6-c10)芳基-n(r”)-(c1-c12)脂族基-,(c6-c10)芳基-(c1-c12)脂族基-o-,(5-10元杂芳基)-(c1-c12)-脂族基-,(5-10元杂芳基)-o-(c1-c12)-脂族基-,(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-,(5-10元杂芳基)-(c1-c12)-脂族基-o-,(3-10元杂环基)-(c1-c12)脂族基-,(3-10元杂环基)-o-(c1-c12)脂族基-,(3-10元杂环基)-n(r”)-(c1-c12)脂族基-和(3-10元杂环基)-(c1-c12)脂族基-o-,其中r2独立地被0-5个r’取代。
674、在式i,ii或iii的化合物的某些实施方案中,r2选自:-h,-me,-et,丙基,异丙基,丁基,叔丁基,环丙基,环丁基,环戊基,环己基,-cf3,-c(o)ome,-c(o)oet,-ome,-ch2ome,-ch2oet,-ch2oph,-ch2-吡咯烷,-ch2-吗啉,-ch2-吡啶和-ch2ph,其中所述r2被0-3个r’取代。在式i,ii或iii的化合物的某些实施方案中,r2是被0-3个r’取代的-me,所述r’选自-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2,其中r”独立地选自h,-(c1-c6)-烷基,(c6-c10)-芳基-和(c6-c10)-芳基-(c1-c6)-烷基-。在某些实施方案中,r2是独立地被0-3个r’取代的-me,所述r’选自-n(me)2,-n(et)2和-n(me)(ch2ph)。
675、在式i,ii或iii的化合物的某些实施方案中,r2选自:-ch2ph,-ch2ch2ph,-ph,-och2ph,-ch2oph,-och2ch2ph,-ch2ch2oph,-ch2-吡咯烷,-ch2-吗啉,-ch2-吡啶和-ch2ph,其中所述ph,吡咯烷,吡啶或吗啉被0-5个r’取代。在式i,ii或iii的化合物的某些实施方案中,r2选自:-ch2ph,-ch2ch2ph,-ph,-och2ph,-ch2oph,-och2ch2ph,-ch2ch2oph,-ch2-吡咯烷,-ch2-吗啉,-ch2-吡啶和-ch2ph,其中所述ph,吡咯烷,吡啶或吗啉被0-5个r’取代,所述r’独立地选自卤素,(c1-c6)-烷基,-oh,-o((c1-c6)-烷基),-ch2oh,-ch2o(c1-c6)-烷基),-ch2n(c1-c6)-烷基)2,-c(o)o(c1-c6)-烷基),-c(o)n(c1-c6)-烷基)2,-no2,-cn,-cf3,-ocf3和-n(c1-c6)-烷基)2。在以上实施方案中的某些中,r2的-ph,吡咯烷,吡啶或吗啉被0-5个r’取代,所述r’独立地选自-f,-cl,-cn,-me,-et,-ome和-oet。在式i,ii或iii的化合物的某些实施方案中,r2是-ch2ph,-ch2oph,-ch2-吡啶,-ch2-吡咯烷或-ch2-吗啉,其中所述-ph,吡咯烷,吡啶或吗啉被0-3个独立地选自-f,-cl,-cn,-me和-ome的r’取代。
676、在式iv的化合物的某些实施方案中,r2是-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8或-(ch2)nn(r”)r10,其中每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;n是选自0-4的整数;p是选自2-4的整数;且每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代。在某些实施方案中,r2是or8。在某些实施方案中,r2是or8,其中r8是被0-5个r’取代的(c6-c10)-芳基。在某些实施方案中,r2是or8,其中r8是被0-3个卤素(诸如-f)取代的(c6-c10)-芳基。在某些实施方案中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8。在某些实施方案中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8是-(c1-c6)烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代。
677、在式i,ii,iii或iv的化合物的某些实施方案中,r3选自:卤素,-r,-cn,-cf3,-so2r,-c(o)n(r)2,-c(o)r和-c(o)or,其中r的每次出现独立地被0-5个r’取代。在某些实施方案中,r3选自:-f,-br,-cl,-(c1-c6)烷基,-cn,-c≡c,-cf3,-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2,-c(o)nh2,-c(o)((c1-c6)烷基),-so2((c6-c10)-芳基),-c(o)o((c1-c6)烷基),-(c2-c6)-烯基,-(c2-c6)-炔基,-(c6-c10)-芳基,5-10元杂芳基-和3-10元杂环基-,其中所述烷基,烯基,炔基,芳基,杂芳基或杂环基-独立地被0-5个r’取代。在式i,ii,iii或iv的化合物的某些实施方案中,r3选自:-h,-c(o)ome,-c(o)et,-c(o)nme2,-c(o)nh2,-c(o)oet,-c(o)och2(叔丁基),-c(o)och2cf3,-c(o)o(异丙基),-c(o)net2,-chf2,-cn,-c≡c,-so2me,-so2et,-so2ph(me),-cf3,-chf2,-me,-et,-br,-cl,-ch2ph,
678、
679、其中r9选自-h,-me,-et,-cf3,异丙基,-ome,-oet,-o-异丙基,-ch2nme2,-叔丁基和环丙基。
680、在式i,ii,iii或iv的化合物的某些实施方案中,r3是-c(o)ome或-c(o)oet。在式i,ii,iii或iv的化合物的某些实施方案中,r3是其中r9选自-h,-me,-et,-cf3,异丙基,-ome,-oet,-o-异丙基,-ch2nme2,-叔丁基和环丙基。
681、在式i,ii,iii或iv的化合物的某些实施方案中,r4和r5各自独立地选自-h,卤素和-r,其中r的每次出现独立地被0-5个r’取代,或者r4和r5可以与它们所结合的碳原子一起形成具有0-3个独立地选自n,o,s,so和so2的另外杂原子的3-10元芳族或非芳族环,其中所述环被0-5个r’取代。在某些实施方案中,r4和r5各自独立地选自-h,-me,-et,-f,或r4和r5与它们所结合的碳原子一起形成3-8元脂族环。在某些实施方案中,r4和r5都是-h。
682、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
683、
684、其中:
685、m是0-3(例如,m是1);
686、每个r1独立地选自:-cl,-f,-ome和-c≡ch;
687、r2是卤素,-(cr2)1-3-or或-(cr2)1-3-o(cr2)1-3-r,其中r的每次出现独立地选自-h,-(c1-c6)烷基,(c6-c10)-芳基-(例如苯基),或5-10元杂芳基-(例如吡啶基)和(c6-c10)-芳基-(c1-c12)脂族基-(例如苯基-(c1-c6)烷基-),且其中r的每次出现独立地被0-5个r’取代;
688、r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-c≡c-苯基,-coome,-cooet,-(c1-c6)烷基,
689、
690、其中r3被0-5个r’取代;
691、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
692、每个r6独立地是-h或-(c1-c6)烷基;
693、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
694、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-,或(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。
695、在某些上述实施方式中,r1是-cl。
696、在某些上述实施方式中,r3选自:
697、
698、其中rt的每次出现独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。在某些实施方式中,r3选自:
699、
700、其中rt的每次出现独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-,和
701、r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基或-异丙基),其中r2独立地被0-5个r’取代。
702、在某些上述实施方式中,r3选自:
703、
704、在某些上述实施方式中,r3选自:
705、
706、和r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基,或-异丙基)。
707、在某些上述实施方式中,r3选自:
708、
709、在某些实施方式中,r3选自:
710、
711、和r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基或-异丙基)。
712、在某些实施方式中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)且其中r2独立地被0-5个r’取代。在某些实施方式中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)且其中r2独立地被0-5个r’取代,和r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-coome,-cooet,-(c1-c6)烷基,其中r3被0-3个r’取代。
713、在某些实施方式中,r2是-ch2or8或-ch2och2r8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)且其中r2独立地被0-5个r’取代;和r3选自:-c≡ch,-c≡c-(c1-c6)烷基,其中r3被0-2个r’取代(例如r3是未经取代的)。
714、在某些实施方式中,本发明提供式ii化合物:
715、
716、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
717、m是0-3(例如m是1);
718、每个r1独立地选自:-cl,-f,-ome,和-c≡ch;
719、r2是卤素或-(cr2)1-3-or,其中r的每次出现独立地选自-h,-(c1-c6)烷基,(c6-c10)-芳基-(例如苯基)和(c6-c10)-芳基-(c1-c12)脂族基-(例如苯基-(c1-c6)烷基-),且其中r的每次出现独立地被0-5个r’取代;
720、r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-c≡c-苯基,
721、其中r3被0-5个r’取代;
722、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
723、每个r6独立地是-h或-(c1-c6)烷基;
724、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
725、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-或(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
726、在以上实施方案中的某些中,r3选自:
727、
728、其中r”的每次出现独立地选自-(c1-c6)-烷基(例如,直链或支链),-c≡ch,苯基,噻吩,(5-10元杂芳基)-(c1-c6)-烷基-和(c6-c10)-芳基-(c1-c6)-烷基-,其中每个r”独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
729、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
730、
731、其中:
732、m是0-3;
733、每个r1独立地选自:卤素(例如,cl,f),-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基)(例如,-ome),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
734、r2选自:-h,卤素,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-c(o)o((c1-c6)烷基),-c(o)nr2,(c6-c10)-芳基-(例如苯基),(c6-c10)-芳基-(c1-c12)脂族基-,(c6-c10)-芳基-o-(c1-c12)脂族基-,(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,(5-10元杂芳基)-(c1-c12)脂族基-,(5-10元杂芳基)-o-(c1-c12)脂族基-,(5-10元杂芳基)-n(r”)-(c1-c12)脂族基-,(3-10元杂环基)-(c1-c12)脂族基-,(3-10元杂环基)-o-(c1-c12)脂族基-和(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
735、其中r2独立地被0-5个r’取代;
736、r3选自:-(c1-c6)烷基,-(c2-c6)烯基(例如,-ch=ch2),-c≡ch,-cn,卤素(例如,br),-so2((c6-c10)-芳基),-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2,-c(o)nh2,-c(o)o((c1-c6)烷基),-c(o)((c1-c6)烷基),-(c6-c10)芳基,5-10元杂芳基(例如,5元杂芳基诸如任选地被取代的)和5-10元杂环基(例如,5元杂环基诸如任选地被取代的),其中r3独立地被0-5个r’取代;
737、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
738、r6选自-h和-(c1-c6)烷基;
739、每个r独立地选自:
740、h-,
741、(c1-c12)-脂族基-,
742、(c3-c10)-环烷基-,
743、(c3-c10)-环烯基-,
744、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
745、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
746、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
747、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
748、(c6-c10)-芳基-,
749、(c6-c10)-芳基-(c1-c12)脂族基-,
750、(c6-c10)-芳基-o-(c1-c12)脂族基-,
751、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
752、3-10元杂环基-,
753、(3-10元杂环基)-(c1-c12)脂族基-,
754、(3-10元杂环基)-o-(c1-c12)脂族基-,
755、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
756、5-10元杂芳基-,
757、(5-10元杂芳基)-(c1-c12)-脂族基-,
758、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
759、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
760、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
761、其中r的每次出现独立地被0-5个r’取代;
762、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
763、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
764、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
765、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
766、
767、其中:
768、m是0-3;
769、每个r1独立地选自:卤素(例如,cl,f),-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基)(例如,-ome),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
770、r2选自:-h,-c(o)nr2和(c6-c10)-芳基-(例如苯基);
771、r3选自:-(c1-c6)烷基,-(c2-c6)烯基(例如,-ch=ch2),-c≡ch,-cn,卤素(例如,br),-so2((c6-c10)-芳基),-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2,-c(o)nh2,-c(o)o((c1-c6)烷基),-c(o)((c1-c6)烷基),-(c6-c10)芳基,5-10元杂芳基(例如,5元杂芳基诸如任选地被取代的)和5-10元杂环基(例如,5元杂环基诸如任选地被取代的),其中r3独立地被0-5个r’取代;
772、r4和r5各自是-h,卤素和-(c1-c6)烷基;
773、r6选自-h和-(c1-c6)烷基;
774、每个r独立地选自:
775、h-,
776、(c1-c12)-脂族基-,
777、(c3-c10)-环烷基-,
778、(c3-c10)-环烯基-,
779、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
780、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
781、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
782、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
783、(c6-c10)-芳基-,
784、(c6-c10)-芳基-(c1-c12)脂族基-,
785、(c6-c10)-芳基-o-(c1-c12)脂族基-,
786、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
787、3-10元杂环基-,
788、(3-10元杂环基)-(c1-c12)脂族基-,
789、(3-10元杂环基)-o-(c1-c12)脂族基-,
790、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
791、5-10元杂芳基-,
792、(5-10元杂芳基)-(c1-c12)-脂族基-,
793、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
794、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
795、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
796、其中r的每次出现独立地被0-5个r’取代;
797、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
798、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
799、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
800、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
801、
802、其中:
803、m是0-3;
804、每个r1独立地选自:卤素(例如,cl,f)和-o((c1-c6)烷基)(例如,-ome),其中r1独立地被0-5个r’取代;
805、r2选自:-h,-c(o)nr2和(c6-c10)-芳基-(例如苯基);
806、r3选自:卤素(例如,br),5-10元杂芳基(例如,5元杂芳基诸如任选地被取代的)和5-10元杂环基(例如,5元杂环基诸如任选地被取代的),其中r3独立地被0-5个r’取代;
807、r4和r5各自是-h;
808、r6是-h;
809、每个r独立地选自:
810、h-,
811、(c1-c12)-脂族基-,
812、(c3-c10)-环烷基-,
813、(c3-c10)-环烯基-,
814、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
815、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
816、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
817、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
818、(c6-c10)-芳基-,
819、(c6-c10)-芳基-(c1-c12)脂族基-,
820、(c6-c10)-芳基-o-(c1-c12)脂族基-,
821、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
822、3-10元杂环基-,
823、(3-10元杂环基)-(c1-c12)脂族基-,
824、(3-10元杂环基)-o-(c1-c12)脂族基-,
825、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
826、5-10元杂芳基-,
827、(5-10元杂芳基)-(c1-c12)-脂族基-,
828、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
829、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
830、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
831、其中r的每次出现独立地被0-5个r’取代;
832、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
833、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
834、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
835、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
836、
837、其中:
838、m是0-3;
839、每个r1独立地选自:卤素(例如,cl,f),-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基)(例如,-ome),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
840、r2选自:-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-c(o)o((c1-c6)烷基),-c(o)nr2,(c6-c10)-芳基-(c6-c10)-芳基-(c1-c12)脂族基-,(c6-c10)-芳基-o-(c1-c12)脂族基-,(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,(5-10元杂芳基)-(c1-c12)脂族基-,(5-10元杂芳基)-o-(c1-c12)脂族基-,(5-10元杂芳基)-n(r”)-(c1-c12)脂族基-,(3-10元杂环基)-(c1-c12)脂族基-,(3-10元杂环基)-o-(c1-c12)脂族基-和(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
841、其中r2独立地被0-5个r’取代;
842、r3选自:-(c2-c6)烯基(例如,-ch=ch2)和5-10元杂环基(例如,5元杂环基诸如任选地被取代的),其中r3独立地被0-5个r’取代;
843、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
844、r6选自-h和-(c1-c6)烷基;
845、每个r独立地选自:
846、h-,
847、(c1-c12)-脂族基-,
848、(c3-c10)-环烷基-,
849、(c3-c10)-环烯基-,
850、[(c3-c10)-环烷基]-(c1-c12)-脂族基-,
851、[(c3-c10)-环烯基]-(c1-c12)-脂族基-,
852、[(c3-c10)-环烷基]-o-(c1-c12)-脂族基-,
853、[(c3-c10)-环烯基]-o-(c1-c12)-脂族基-,
854、(c6-c10)-芳基-,
855、(c6-c10)-芳基-(c1-c12)脂族基-,
856、(c6-c10)-芳基-o-(c1-c12)脂族基-,
857、(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,
858、3-10元杂环基-,
859、(3-10元杂环基)-(c1-c12)脂族基-,
860、(3-10元杂环基)-o-(c1-c12)脂族基-,
861、(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
862、5-10元杂芳基-,
863、(5-10元杂芳基)-(c1-c12)-脂族基-,
864、(5-10元杂芳基)-o-(c1-c12)-脂族基-;和
865、(5-10元杂芳基)-n(r”)-(c1-c12)-脂族基-;
866、其中所述杂环基具有1-4个独立地选自n、nh、o、s、so和so2的杂原子,且所述杂芳基具有1-4个独立地选自n、nh、o和s的杂原子;
867、其中r的每次出现独立地被0-5个r’取代;
868、或当两个r基团结合至相同原子时,所述两个r基团可以与它们所结合的原子一起形成具有0-4个独立地选自n、nh、o、s、so和so2的杂原子的3-10元芳族或非芳族环,其中所述环任选地被0-5个r’取代,且其中所述环任选地与(c6-c10)芳基,5-10元杂芳基,(c3-c10)环烷基或3-10元杂环基稠合;
869、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2n(r”)2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
870、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
871、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
872、
873、其中:
874、m是0-3;
875、每个r1独立地选自:卤素(例如,cl,f)和-o((c1-c6)烷基)(例如,-ome),其中r1独立地被0-5个r’取代;
876、r2选自:-h,-(c1-c6)烷基,(c6-c10)-芳基-(例如苯基)和(c6-c10)-芳基-(c1-c12)脂族基-,
877、其中r2独立地被0-5个r’取代;
878、r3选自:-(c2-c6)烯基(例如,-ch=ch2)和5-10元杂环基(例如,5元杂环基诸如任选地被取代的),其中r3独立地被0-5个r’取代;
879、r4和r5各自是-h;
880、r6是-h;
881、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2n(r”)2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
882、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
883、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
884、
885、其中:
886、m是0-3;
887、每个r1独立地选自:卤素,-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-no2,-cn,-cf3和-ocf3,其中所述烷基独立地被0-5个r’取代;
888、r2选自:-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-c(o)o((c1-c6)烷基),(c6-c10)-芳基-(c1-c12)脂族基-,(c6-c10)-芳基-o-(c1-c12)脂族基-,(c6-c10)-芳基-(c1-c12)脂族基-o-,(3-10元杂环基)-(c1-c12)脂族基-,(5-10元杂芳基)-(c1-c12)-脂族基-,(5-10元杂芳基)-o-(c1-c12)-脂族基-和(5-10元杂芳基)-(c1-c12)-脂族基-o-,其中所述烷基,芳基或杂芳基独立地被0-5个r’取代;
889、r3选自:-(c1-c6)烷基,-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2和-c(o)o((c1-c6)烷基),其中所述烷基独立地被0-5个r’取代;
890、r’如本文中定义;
891、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;且
892、r6选自-h和-(c1-c6)烷基。
893、在式ii的化合物的某些实施方案中,m是0,1或2;
894、当m是1或2时,r1的至少一次出现是卤素或-o((c1-c6)烷基)(诸如-f和-ome);
895、r2选自:-(c1-c6)烷基(例如,-me),(c6-c10)-芳基-(c1-c12)脂族基-(例如,-ch2ph),(c6-c10)-芳基-o-(c1-c12)脂族基-(例如,-ch2oph)和(3-10元杂环基)-(c1-c12)脂族基-(例如,-ch2-吡咯烷和-ch2-吗啉),其中所述芳基(例如,-ph)或杂环基(例如,吡咯烷或吗啉)独立地被0-5个r’取代,所述r’独立地选自-f,-me和-ome,且其中所述烷基(例如,-me)独立地被0-3个选自-n(et)2和-n(me)(ch2ph)的r’取代。
896、r3是-c(o)o((c1-c6)烷基)(例如,-cooet);
897、r4和r5都是-h;且
898、r6是-h。
899、在某些实施方案中,本发明提供了式ii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
900、
901、其中:
902、m是0-3;
903、每个r1独立地选自:卤素,-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
904、r2选自:-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-c(o)o((c1-c6)烷基),(c6-c10)-芳基-(c1-c12)脂族基-,(c6-c10)-芳基-o-(c1-c12)脂族基-,(c6-c10)-芳基-n(r”)-(c1-c12)脂族基-,(5-10元杂芳基)-(c1-c12)脂族基-,(5-10元杂芳基)-o-(c1-c12)脂族基-,(5-10元杂芳基)-n(r”)-(c1-c12)脂族基-,(3-10元杂环基)-(c1-c12)脂族基-,(3-10元杂环基)-o-(c1-c12)脂族基-和(3-10元杂环基)-n(r”)-(c1-c12)脂族基-,
905、其中r2独立地被0-5个r’取代;
906、r3选自:-(c1-c6)烷基,-c≡c,-cn,卤素,-so2((c6-c10)-芳基),-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2,-c(o)nh2,-c(o)o((c1-c6)烷基),-c(o)((c1-c6)烷基),-(c6-c10)芳基和5-10元杂芳基,其中r3独立地被0-5个r’取代;
907、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
908、r6选自-h和-(c1-c6)烷基;且
909、r’和r”如本文中定义。
910、在式ii的化合物的某些实施方案中:m是0,1或2;
911、当m是1或2时,r1的至少一次出现是卤素或-o((c1-c6)烷基);
912、r2选自:
913、-(c1-c6)烷基,(c6-c10)-芳基-(c1-c12)脂族基-,(c6-c10)芳基-o-(c1-c12)脂族基-,(5-10元杂芳基)-(c1-c12)脂族基-和(3-10元杂环基)-(c1-c12)脂族基-,其中r2独立地被0-3个r’取代;
914、r3是卤素,-cn,-c≡c,-c(o)nh2,-(c1-c6)烷基,-c(o)((c1-c6)烷基),-c(o)o((c1-c6)烷基),-so2(ph(me)),
915、其中r3独立地被0-3个r’取代,且其中r9选自-h,-me,-et,-cf3,异丙基,-ome,-叔丁基和环丙基;
916、r4和r5都是-h;
917、r6是-h;且
918、r’如本文中定义。
919、在式ii的化合物的某些实施方案中,r3是:其中r9选自-h,-me,-et,-cf3,异丙基,-ome和-叔丁基。
920、在某些实施方案中,本发明提供了式iii的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
921、
922、其中:
923、m是0,1或2,且当m是1或2时,r1的至少一次出现是-o((c1-c6)烷基)(诸如-ome);
924、r2选自:-(c1-c6)烷基(例如,-me)和(c6-c10)-芳基-(c1-c12)脂族基-(例如,-ch2ph);
925、r3是-c(o)o((c1-c6)烷基)(例如,-cooet);
926、r4和r5都是-h;且
927、r6是-h。
928、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
929、
930、iv,
931、或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合,其中:
932、m是0-3(例如m是1);
933、每个r1独立地选自:-cl,-f,-ome,和-c≡ch;
934、r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是-(c1-c6)烷基,(c6-c10)-芳基(例如苯基),或5-10元杂芳基-(例如吡啶基)和其中r2独立地被0-5个r’取代;
935、r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-c≡c-苯基,-coome,-cooet,-(c1-c6)烷基,其中r3被0-5个r’取代;
936、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
937、每个r6独立地是-h或-(c1-c6)烷基;
938、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
939、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-,和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个rt取代,所述rt独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。
940、在某些上述实施方式中,r1是-cl。
941、在某些上述实施方式中,r3选自:
942、
943、其中rt的每次出现独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-。在某些实施方式中,r3选自:
944、
945、其中rt的每次出现独立地选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,和(c6-c10)-芳基-,和
946、r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基或-异丙基),其中r2独立地被0-5个r’取代。
947、在某些上述实施方式中,r3选自:
948、
949、在某些实施方式中,r3选自:
950、
951、和r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基,或-异丙基)。
952、在某些上述实施方式中,r3选自:
953、
954、在某些实施方式中,r3选自:
955、
956、和r2是-(ch2)nor8,其中r8是-(c1-c6)烷基(例如-me,-et,-丙基,或-异丙基)。
957、在某些实施方式中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)和其中r2独立地被0-5个r’取代。在某些实施方式中,r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)和其中r2独立地被0-5个r’取代,和r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-coome,-cooet,-(c1-c6)烷基,其中r3被0-3个r’取代。
958、在某些实施方式中,r2是-ch2or8或-ch2och2r8,其中r8的每次出现独立地是(c6-c10)-芳基(例如苯基)或5-10元杂芳基-(例如吡啶基)和其中r2独立地被0-5个r’取代;和r3选自:-c≡ch,-c≡c-(c1-c6)烷基,其中r3被0-2个r’取代(例如r3是未经取代的)。
959、在又一方面,本发明提供式iv化合物:
960、
961、其中:
962、m是0-3(例如,m是1);
963、每个r1独立地选自:-cl,-f,-ome和-c≡ch;
964、r2是-(ch2)nor8或-(ch2)no(ch2)nr8,其中r8的每次出现独立地是-(c1-c6)烷基或(c6-c10)-芳基(例如苯基),且其中r2独立地被0-5个r’取代;
965、r3选自:-cn,-c≡ch,-c≡c-(c1-c6)烷基,-c≡c-苯基,
966、其中r3被0-5个r’取代;
967、r4和r5的每次出现独立地是-h或-(c1-c6)烷基;
968、每个r6独立地是-h或-(c1-c6)烷基;
969、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
970、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-,其中r”的每次出现独立地被0-5个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
971、在以上实施方案中的某些中,r3选自:
972、
973、其中r”的每次出现独立地选自-(c1-c6)-烷基(例如,直链或支链),-c≡ch,苯基,噻吩,(5-10元杂芳基)-(c1-c6)-烷基-和(c6-c10)-芳基-(c1-c6)-烷基-,其中每个r”独立地被0-3个取代基取代,所述取代基选自:卤素,-ro,-oro,氧代,-ch2oro,-ch2n(ro)2,-c(o)n(ro)2,-c(o)oro,-no2,-ncs,-cn,-cf3,-ocf3和-n(ro)2,其中ro的每次出现独立地选自:-(c1-c6)-脂族基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-和(c6-c10)-芳基-。
974、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
975、
976、其中:
977、m是0-3;
978、每个r1独立地选自:卤素(例如,cl),-h,-(c1-c6)烷基,-c≡ch,-oh,-o((c1-c6)烷基)(例如,ome),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
979、r2选自-or8,-sr8,-(ch2)nor8(例如,-ch2ome,-ch2oet,-ch2o异丙基,-ch2o吡啶基),-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10,其中n是选自0-4的整数;p是选自2-4的整数;每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;且其中r2独立地被0-5个r’取代;
980、r3选自:-h,-cn,卤素(例如,br),-(c1-c6)烷基,-c≡ch,-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2),-c(o)nh((c1-c6)脂族基)2(例如,-c(o)nh((c2-c6)炔基)2),(c6-c10)-芳基-(c1-c12)脂族基-,-c(o)((c1-c6)烷基),-c(o)o((c1-c6)烷基),5或6元杂环基-(例如,任选地被取代的或任选地被取代的)和5或6元杂芳基(例如,任选地被取代的任选地被取代的其中r9选自-me,-et,异丙基,-cf3,-ome,-oet,-o-异丙基,-ch2nme2和环丙基;且其中r3独立地被0-5个r’取代;
981、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
982、r6选自-h和-(c1-c6)烷基;
983、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2n(r”)2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
984、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
985、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
986、
987、其中:
988、m是1;
989、r1是任选地被r’取代的-c≡ch;
990、r2选自-or8,-sr8,-(ch2)nor8(例如,-ch2ome,-ch2oet,-ch2o异丙基,-ch2o吡啶基),-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10,其中n是选自0-4的整数;p是选自2-4的整数;每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;且其中r2独立地被0-5个r’取代;
991、r3选自:-h,-cn,卤素(例如,br),-(c1-c6)烷基,-c≡ch,-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2),-c(o)nh((c1-c6)脂族基)2(例如,-c(o)nh((c1-c6)炔基)2),(c6-c10)-芳基-(c1-c12)脂族基-,-c(o)((c1-c6)烷基),-c(o)o((c1-c6)烷基),5或6元杂环基-(例如,任选地被取代的或任选地被取代的)和5或6元杂芳基(例如,任选地被取代的任选地被取代的其中r9选自-me,-et,异丙基,-cf3,-ome,-oet,-o-异丙基,-ch2nme2和环丙基;且其中r3独立地被0-5个r’取代;
992、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
993、r6选自-h和-(c1-c6)烷基;
994、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
995、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
996、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
997、
998、其中:
999、m是1;
1000、每个r1是任选地被r’取代的-c≡ch;
1001、r2是-(ch2)nor8(例如,-ch2ome,-ch2oet,-ch2o异丙基,-ch2o吡啶基);且其中r2独立地被0-5个r’取代;
1002、r3选自:5或6元杂环基-(例如,任选地被取代的或任选地被取代的)和5或6元杂芳基(例如,任选地被取代的或任选地取代的);且其中r3独立地被0-5个r’取代;
1003、r4和r5各自是-h;
1004、r6是-h;且
1005、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
1006、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
1007、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
1008、
1009、其中:
1010、m是0-3;
1011、当m是1或2时,r1的至少一次出现是-卤素或-o((c1-c6)烷基);
1012、每个r1独立地选自:卤素(例如,cl),-h,-(c1-c6)烷基,-c≡ch,-oh,-o((c1-c6)烷基)(例如,ome),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
1013、r2选自-or8,-sr8,-(ch2)nor8(例如,-ch2ome,-ch2oet,-ch2o异丙基,-ch2o吡啶基),-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10,其中n是选自0-4的整数;p是选自2-4的整数;每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;且其中r2独立地被0-5个r’取代;
1014、r3选自:-c≡ch,-c(o)nh((c1-c6)脂族基)2(例如,-c(o)nh((c1-c6)炔基)2),(c6-c10)-芳基-(c1-c12)脂族基-,5或6元杂环基-(例如,任选地被取代的或任选地被取代的),任选地被取代的和任选地被取代的且其中r3独立地被0-5个r’取代;
1015、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
1016、r6选自-h和-(c1-c6)烷基;且
1017、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
1018、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
1019、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
1020、
1021、其中:
1022、m是0-3;
1023、每个r1独立地选自:卤素(例如,cl),-c≡ch和-o((c1-c6)烷基)(例如,ome),其中r1独立地被0-5个r’取代;
1024、r2是-(ch2)nor8(例如,-ch2ome,-ch2oet,-ch2o-异丙基,-ch2o-吡啶基),其中n是选自0-4的整数;r8是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;且其中r2独立地被0-5个r’取代;
1025、r3选自:-c≡ch,-c(o)nh((c1-c6)脂族基)2(例如,-c(o)nh((c1-c6)炔基)2)),(c6-c10)-芳基-(c1-c12)脂族基-,5或6元杂环基-(例如,任选地被取代的或任选地被取代的),任选地被取代的和任选地被取代的且其中r3独立地被0-5个r’取代;
1026、r4和r5各自是-h;
1027、r6是-h或-(c1-c6)烷基;且
1028、其中r’的每次出现独立地选自卤素,-r”,-or”,氧代,-ch2or”,-ch2nr”2,-c(o)n(r”)2,-c(o)or”,-no2,-ncs,-cn,-cf3,-ocf3和-n(r”)2;
1029、其中r”的每次出现独立地选自h,-(c1-c6)-烷基,(c3-c6)-环烷基,3-6元杂环基,5-10元杂芳基-,(c6-c10)-芳基-,(5-10元杂芳基)-(c1-c6)-烷基-,(c6-c10)-芳基-(c1-c6)-烷基-,(5-10元杂芳基)-o-(c1-c6)-烷基-和(c6-c10)-芳基-o-(c1-c6)-烷基-。
1030、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
1031、
1032、其中:
1033、m是0,1或2,且当m是1或2时,r1的至少一次出现是-o((c1-c6)烷基)(诸如-ome);
1034、r2是or8,其中r8是被0-3个卤素(诸如-f)取代的(c6-c10)-芳基(诸如苯基);
1035、r3是-c(o)o((c1-c6)烷基)(例如,-cooet);
1036、r4和r5都是-h;且
1037、r6是-h。
1038、在另一个方面,本发明提供了式iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合:
1039、
1040、其中:
1041、m是0-3;
1042、当m是1或2时,r1的至少一次出现是-卤素或-o((c1-c6)烷基);
1043、每个r1独立地选自:卤素,-h,-(c1-c6)烷基,-oh,-o((c1-c6)烷基),-no2,-cn,-cf3和-ocf3,其中r1独立地被0-5个r’取代;
1044、r2选自-or8,-sr8,-(ch2)nor8,-(ch2)no(ch2)nr8,-(ch2)pr8和-(ch2)nn(r”)r10,其中n是选自0-4的整数;p是选自2-4的整数;每个r8独立地是-(c1-c6)烷基,-(c3-c10)-环烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8的每次出现独立地被0-5个r’取代;每个r10独立地是-(c3-c10)-环烷基,3-10元杂环基-,(c6-c10)-芳基或5-10元杂芳基,其中r10的每次出现独立地被0-5个r’取代;且其中r2独立地被0-5个r’取代;
1045、r3选自:-h,-cn,卤素,-(c1-c6)烷基,-so2((c1-c6)烷基),-c(o)n((c1-c6)烷基)2,-c(o)((c1-c6)烷基),-c(o)o((c1-c6)烷基),其中r9选自-me,-et,异丙基,-cf3,-ome,-oet,-o-异丙基,-ch2nme2和环丙基;且其中r3独立地被0-5个r’取代;
1046、r4和r5各自独立地选自-h,卤素和-(c1-c6)烷基;
1047、r6选自-h和-(c1-c6)烷基;且
1048、r’和r”如本文中定义。
1049、在式iv的化合物的某些实施方案中:
1050、m是0,1或2;
1051、r2是-or8,-(ch2)nor8,-(ch2)no(ch2)nr8,其中n是1,且其中r8是-(c1-c6)烷基,(c6-c10)-芳基或5-10元杂芳基,其中r8独立地被0-3个r’取代;
1052、r3是卤素,-h,-cn,-(c1-c6)烷基,-c(o)((c1-c6)烷基),-c(o)o((c1-c6)烷基),其中所述烷基独立地被0-3个r’取代;r9选自-me,-et,异丙基和-cf3;
1053、r4和r5都是-h;
1054、r6是-h;且
1055、r’如本文中定义。
1056、本技术的具体化合物的例子包括:
1057、
1058、
1059、
1060、
1061、
1062、
1063、
1064、
1065、
1066、
1067、
1068、
1069、
1070、
1071、
1072、
1073、
1074、
1075、
1076、
1077、
1078、
1079、
1080、
1081、
1082、
1083、
1084、
1085、
1086、
1087、
1088、
1089、
1090、
1091、
1092、
1093、
1094、
1095、
1096、
1097、
1098、
1099、
1100、
1101、
1102、
1103、
1104、
1105、
1106、
1107、
1108、
1109、
1110、
1111、
1112、
1113、
1114、
1115、
1116、
1117、
1118、
1119、
1120、
1121、
1122、和它们的药学上合适的盐,水合物,溶剂化物,多晶型物,异构体或它们的组合。
1123、本发明还包括如上文所述的r1,r2和r3的多种组合。这些组合又可以与本文所述的其它变量的任意或所有值组合。例如,r1可以是-or或卤素;r2可以是(c1-c4)-烷基-,-or8,-(ch2)nor8或-(ch2)no(ch2)nr8;且任选地r3是-c(o)or或-c(o)n(r)2。在另一个实施例中,r1是-or或卤素;r2是(c1-c4)-烷基-,-or8,-(ch2)nor8或-(ch2)no(ch2)nr8;且r3是5或6元杂芳基,诸如对于以上实施例中的每一个,化合物可以具有本文所述基团的特定值。
1124、除非另外指出,否则本文描述的任意实施方案还意欲表示化合物的未标记的形式以及同位素标记的形式。同位素标记的化合物具有本文给出的结构式所描绘的结构,但是一个或多个原子被具有所选择的原子质量或质量数的原子替代。可以掺入本发明的化合物中的同位素的例子包括氢,碳,氮,氧,磷,氟和氯的同位素,分别诸如2h,3h,11c,13c,14c,15n,18f,31p,32p,35s,36cl,125i。本发明包括各种同位素标记的本文所定义的化合物,例如其中存在放射性同位素诸如3h,13c和14c的那些。这样的同位素标记的化合物可用于代谢研究(优选使用14c),反应动力学研究(使用例如2h或3h),检测或成像技术,诸如正电子发射断层成像术(pet)或单光子发射计算机断层成像术(spect),包括药物或底物组织分布测定或在患者的放射性治疗中。特别地,对pet或spect研究而言特别优选18f或标记的化合物。一般可以通过实施下述方案或实施例和制备中所公开的方法,用易于得到的同位素标记的试剂替代非同位素标记的试剂,制备同位素标记的本发明的化合物及其前药。
1125、本文列出的各个实施方案中的任一个可以分别定义式i,ii,iii,iv,v,vi,vii,viii或ix或可以组合以产生本发明的优选实施方案。
1126、通用合成方法
1127、一般而言可以通过本领域技术人员已知的方法制备本发明的化合物。如下方案1-10提供了制备式i-iv的化合物的通用合成途径。对普通熟练有机化学家显而易见的其它等效方案可以可替换地用于合成下列通用方案所示例的分子的不同部分。
1128、方案1.式i的化合物(其中x,y,z,v和w形成1,2,3-三唑环)或式ii的化合物的通用合成。
1129、
1130、方案2.式i或iii的化合物(其中x,y,z,v和w形成吡唑环)的通用合成。
1131、
1132、方案3.式i的化合物(其中x,y,z,v和w形成苯氧基-取代的1,2,3-三唑环)或式ii的化合物的通用合成。
1133、
1134、方案4.式i或ii的化合物的通用合成,以允许在由x,y,z,v和w形成的三唑并-环上的发散性官能团化。
1135、
1136、方案5.式i的化合物(其中x,y,z,v和w形成氨基甲基-取代的1,2,3-三唑环)或式ii的化合物的通用合成。
1137、
1138、方案6.式i的化合物(其中x,y,z,v和w形成芳烷基-取代的或杂芳烷基取代的1,2,3-三唑环)或式ii的化合物的通用合成。
1139、
1140、方案7.式i或iv的化合物(其中x,y,z,v和w形成被取代的1,2,4-三唑环)的通用合成。
1141、
1142、方案8.式i的化合物(其中x,y,z,v和w形成甲基-取代的1,2,3-三唑环)或式ii的化合物的通用合成。
1143、
1144、方案9.式i的化合物(其中x,y,z,v和w形成苄基-取代的1,2,3-三唑环)或式ii的化合物的通用合成。
1145、
1146、方案10.式i,ii或iv化合物的通用合成,其中x,y,z,v和w形成被取代的三唑环,诸如1,2,3-三唑环或1,2,4-三唑环,且上面的咪唑被如10(a)和10(b)中所示的1,2,4-噁二唑环取代。
1147、
1148、方案10a.在方案10a中示例了化合物(其中r3是任选地被取代的二氢噁唑或噁嗪基环)的通用合成。
1149、
1150、方案10b(a)和10b(b).在方案10b(a)和10b(b)中示例了化合物(其中r3是任选地被取代的噁唑或异噁唑)的通用合成。
1151、(a)
1152、(b)
1153、方案10c.在方案10c中示例了化合物(其中r3是任选地被取代的炔基)的通用合成。
1154、
1155、正如熟练的从业人员认识到的,通过改变化学试剂或合成途径可以制备具有不同于上文所述那些的变量的式i-iv的化合物。
1156、药物组合物和施用模式
1157、本发明提供了药物组合物,其包含药学上可接受的载体和式i-iv的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1158、可以用诸如以下试剂使存在于本发明的化合物中的碱性含氮基团季铵化:低级烷基卤,诸如甲基,乙基,丙基和丁基的氯化物,溴化物和碘化物;二烷基硫酸酯,诸如二甲基,二乙基,二丁基和二戊基硫酸酯;长链卤化物,诸如癸基,月桂基,肉豆蔻基和硬脂酰基的氯化物,溴化物和碘化物;芳烷基卤,诸如苄基和苯乙基的溴化物等。由此获得水或油可溶性的或可分散的产物。
1159、应当理解,用于本发明的组合物中的化合物和试剂优选地在外周施用时应当容易穿透血脑屏障。然而,不可穿透血脑屏障的化合物仍可以有效地直接施用进中枢神经系统中,例如,通过脑室内(intraventricular)或其它神经-相容的途径。
1160、在本发明的某些实施方案中,将含有α5的gabaa r正变构调节剂与药学上可接受的载体一起配制。可以用在这些组合物中的药学上可接受的载体包括但不限于:离子交换剂,氧化铝,硬脂酸铝,卵磷脂,血清蛋白,诸如人血清白蛋白,缓冲物质,诸如磷酸盐,甘氨酸,山梨酸,山梨酸钾,饱和植物脂肪酸的偏甘油酯混合物,水,盐或电解质,诸如硫酸鱼精蛋白,磷酸氢二钠,磷酸氢钾,氯化钠,锌盐,胶态二氧化硅,三硅酸镁,聚乙烯吡咯烷酮,基于纤维素的物质,聚乙二醇,羧甲纤维素钠,聚丙烯酸酯,蜡类,聚乙烯-聚氧丙烯-嵌段共聚物,聚乙二醇和羊毛脂。在其它实施方案中,没有使用载体。例如,含有α5的gabaa r激动剂(例如,含有α5的gabaa受体正变构调节剂)可以单独施用或作为药物制剂(治疗组合物)的组分施用。可以配制含有α5的gabaa r激动剂(例如,含有α5的gabaa受体正变构调节剂)用于以用在人药物中的任意便利的方式施用。
1161、在某些实施方案中,本发明的治疗方法包括表面地(topically),全身地或局部地(locally)施用化合物或试剂的组合物。例如,可以配制本发明的化合物或试剂的治疗组合物用于通过以下方式施用,例如,注射(例如静脉内,皮下或肌内),吸入或吹入(通过口或鼻)或口服,含服,舌下,透皮,鼻部或胃肠外施用。可以将本文所述的化合物或试剂的组合物配制为植入物或装置的组成部分,或配制用于缓释或延长释放。当胃肠外地施用时,用于本发明中的化合物或试剂的治疗组合物优选地是无热原的生理学可接受的形式。技术和制剂一般可以在remington’spharmaceutical sciences,meade publishing co.,easton,pa中找到。
1162、在某些实施方案中,适用于胃肠外施用的药物组合物可以包含含有α5的gabaa r正变构调节剂和与其组合的一种或多种药学上可接受的无菌的等渗的水性的或非水性的溶液,分散体(dispersion),悬浮液或乳剂或无菌粉剂,所述粉剂可以在临用前重构成无菌的可注射的溶液或分散体,其可含有抗氧化剂,缓冲剂,抑菌剂,使所述制剂与预定的受体的血液等渗的溶质或者助悬剂或增稠剂。可以用于本发明的药物组合物中的合适的水性和非水性载体的例子包括水,乙醇,多元醇(如甘油,丙二醇,聚乙二醇等)及其适当的混合物,植物油,诸如橄榄油,以及可注射的有机酯,如油酸乙酯。可以维持适当的流动性,例如,通过使用包衣材料如卵磷脂,在分散体的情况下通过维持所需粒度,以及通过使用表面活性剂。
1163、包含含有α5的gabaa r正变构调节剂的组合物还可以含有辅助剂,诸如防腐剂,润湿剂,乳化剂和分散剂(dispersing agent)。通过包含各种抗细菌剂和抗真菌剂,例如,对羟基苯甲酸酯,三氯叔丁醇,苯酚山梨酸等,可以确保阻止微生物的作用。也可能需要在组合物中包含等渗剂,诸如糖,氯化钠等。另外,通过包含延迟吸收的试剂诸如单硬脂酸铝和明胶,可以实现可注射药物形式的延长吸收。
1164、在本发明的某些实施方案中,包含含有α5的gabaa r正变构调节剂的组合物可以口服施用,例如,以胶囊剂,扁囊剂,丸剂,片剂,锭剂(使用经调味的基质,通常为蔗糖和阿拉伯胶或黄蓍胶),粉剂,颗粒剂的形式,或者作为在水性或非水性液体中的溶液或悬浮液,或者作为水包油或油包水液体乳剂,或者作为酏剂或糖浆剂,或者作为软锭剂(pastilles)(使用惰性基质,诸如明胶和甘油,或蔗糖和阿拉伯胶)等,各自含有预定量的含有α5的gabaa r正变构调节剂作为活性成分。
1165、在用于口服施用的固体剂型(胶囊剂,片剂,丸剂,糖衣丸,粉剂,颗粒等)中,可以将一种或多种包含含有α5的gabaa r正变构调节剂的组合物与一种或多种药学上可接受的载体(诸如柠檬酸钠或磷酸二钙)和/或以下任一种混合:(1)填充剂或增量剂,诸如淀粉,乳糖,蔗糖,葡萄糖,甘露醇和/或硅酸;(2)粘合剂,例如,羧甲纤维素,海藻酸盐,明胶,聚乙烯吡咯烷酮,蔗糖和/或阿拉伯胶;(3)保湿剂,诸如甘油;(4)崩解剂,诸如琼脂,碳酸钙,马铃薯或木薯淀粉,海藻酸,某些硅酸盐和碳酸钠;(5)溶液阻滞剂,诸如石蜡;(6)吸收促进剂,诸如季铵化合物;(7)润湿剂,例如,鲸蜡醇和单硬脂酸甘油酯;(8)吸收剂,诸如高岭土和皂粘土粘土;(9)润滑剂,诸如滑石,硬脂酸钙,硬脂酸镁,固体聚乙二醇,月桂基硫酸钠,及其混合物;和(10)着色剂。在胶囊剂,片剂和丸剂的情况下,所述药物组合物还可以包含缓冲剂。也可以使用类似类型的固体组合物,作为在软和硬填充的明胶胶囊中的填充剂并使用诸如乳糖(lactose)或牛奶糖(milk sugar)以及高分子量聚乙二醇等的赋形剂。
1166、用于口服施用的液体剂型包括药学上可接受的乳剂,微乳剂,溶液,悬浮液,糖浆剂和酏剂。除含有α5的gabaa r正变构调节剂外,液体剂型还可以含有本领域中常用的惰性稀释剂诸如水或其它溶剂,增溶剂和乳化剂,诸如乙醇(乙醇),异丙醇,碳酸乙酯,乙酸乙酯,苄醇,苯甲酸苄酯,丙二醇,1,3-丁二醇,油(特别是棉籽油,花生油,玉米油,胚油,橄榄油,蓖麻油和芝麻油),甘油,四氢糠醇,聚乙二醇和脱水山梨糖醇的脂肪酸酯,及其混合物。除惰性稀释剂外,口服组合物还可以包含辅助剂诸如润湿剂,乳化剂和助悬剂,甜味剂,矫味剂,着色剂,香味剂和防腐剂。
1167、除活性化合物以外,悬浮液还可以含有助悬剂,诸如乙氧基化的异硬脂醇,聚氧乙烯山梨醇和脱水山梨糖醇酯,微晶纤维素,氢氧化铝氧化物(alumium metahydroxide),皂粘土,琼脂和黄蓍胶,及其混合物。
1168、如本文中所述的,可以施用化合物,试剂及其组合物用于缓释,控释或延长释放。术语"延长释放"是制药科学领域中广泛知道的,且在本文中用于表示活性化合物或试剂在延长的时间段内(诸如大于或等于1小时)(自始至终或过程中)从剂型控释进入环境。延长释放剂型将在延长的时间段内以基本上恒定速率释放药物,或在延长的时间段内以递增的方式释放基本上恒定量的药物。本文中使用的术语"延长释放"包括术语"控释","延时释放(prolonged release)","持续释放","延迟释放"或"缓释",如在制药科学中使用的这些术语。在某些实施方案中,以贴剂或泵的形式施用延长释放剂量。
1169、本领域普通技术人员(诸如医师)易于能够确定使用本发明的组合物和方法治疗受试者所需的含有α5的gabaa r正变构调节剂的量。应当理解,将针对个体确定剂量方案,并考虑例如改变含有α5的gabaa r正变构调节剂的作用的各种因素,疾病的严重程度或阶段,施用途径和个体独有的特征,诸如年龄,体重,大小和认知损害程度。
1170、本领域众所周知,对体表面积归一化是推断物种之间剂量的适合方法。为了从用于治疗大鼠中年龄依赖性认知损害的剂量计算人体等效剂量(hed),可以采用公式hed(mg/kg)=大鼠剂量(mg/kg)×0.16(参见estimating the safe starting dose in clinicaltrials for therapeutics in adult healthy volunteers,2002年12月,center forbiologics evaluation和research)。例如,使用该公式,大鼠中的10mg/kg剂量与人体中的1.6mg/kg等效。这种转化是基于更一般性的公式hed=以mg/kg计的动物剂量×(以kg计的动物体重/以kg计的人体重)0.33。
1171、在本发明的某些实施方案中,含有α5的gabaa r正变构调节剂的剂量是在0.0001至100mg/kg/天之间(给定70kg的典型人受试者,它是在0.007至7000mg/天之间)。
1172、在本发明的某些实施方案中,施用间隔是每12或24小时1次。还可以使用在更低频率间隔的施用诸如每6小时1次。
1173、如果通过植入物,装置或缓释或延长释放制剂施用,则可以将含有α5的gabaa r正变构调节剂施用一次,或在必要时贯穿患者寿命定期施用一次或多次。还可以使用对临床应用而言处于这些剂量间隔中间或更短的其它施用间隔,并且可以由本领域技术人员按照本发明的方法确定。
1174、本领域技术人员可以通过例行实验确定期望的施用时间。例如,可以将含有α5的gabaa r正变构调节剂施用1-4周,1-3个月,3-6个月,6-12个月,1-2年或更多直到患者寿命的时间段。
1175、除含有α5的gabaa r正变构调节剂外,本发明的组合物还可以包括其它治疗上有用的试剂。这些其它治疗上有用的试剂可以根据本发明的方法与含有α5的gabaa r正变构调节剂一起在单一制剂中同时或依次施用。
1176、本领域普通技术人员应该理解,当适合于所致力的应用时,可以修改和修饰本文所述的组合物,且本文所述的组合物可以用于其它适合的应用。例如,本技术的组合物还可以包含第二种治疗剂。这样的其它添加和改变不脱离其范围。
1177、具有抗精神病药的药物组合物
1178、本技术的化合物或组合物可以与抗精神病药联合用于治疗受试者中的与精神分裂症或双相型障碍有关的认知损害,所述受试者具有所述精神分裂症或双相型障碍(例如,躁狂症)或处于其风险中。可用于本发明的方法和组合物中的抗精神病药或其药学上可接受的盐,水合物,溶剂化物或多晶型物包括典型的和非典型抗精神病药。在某些实施方案中,本发明的化合物或组合物可用于治疗一种或多种与精神分裂症相关的阳性的和/或阴性的症状以及认知损害。在某些实施方案中,本发明的化合物或组合物可用于治疗一种或多种与双相型障碍(特别是躁狂症)相关的症状以及认知损害。在本发明的某些实施方案中,本发明的化合物或组合物在所述受试者中预防或减慢精神分裂症或双相型障碍(特别是躁狂症)的认知损害的发展。
1179、在某些实施方案中,适合用在本发明中的抗精神病药选自非典型抗精神病药。这样的非典型抗精神病药包括但不限于在例如美国专利4,734,416,5,006,528,4,145,434,5,763,476,3,539,573,5,229,382,5,532,372,4,879,288,4,804,663,4,710,500,4,831,031和5,312,925和欧洲专利ep402644和ep368388中公开的那些及其药学上可接受的盐,水合物,溶剂化物和多晶型物。
1180、在某些实施方案中,适合用在本发明中的非典型抗精神病药包括但不限于阿立哌唑,阿塞那平,氯氮平,伊潘立酮,奥氮平,鲁拉西酮,帕利哌酮,喹硫平,利培酮和齐拉西酮及其药学上可接受的盐,水合物,溶剂化物和多晶型物。在某些实施方案中,适合用于本文用途的抗精神病药选自阿立哌唑(bristol-myers squibb),奥氮平(lilly)和齐拉西酮(pfizer)及其药学上可接受的盐,水合物,溶剂化物和多晶型物。
1181、在某些实施方案中,适合用在本发明中的抗精神病药是典型的抗精神病药,包括但不限于乙酰丙嗪,苯哌利多,溴西泮,溴哌利多,氯丙嗪,氯普噻吨,氯噻平,氰美马嗪,地西泮,地西拉嗪,氟哌利多,氟哌噻吨,氟奋乃静,氟司必林,氟哌啶醇,辛胺醇,异丙碘铵,左美丙嗪,左舒必利,洛沙平,美哌隆,美索达嗪,吗茚酮,奥昔哌汀,甲羟哌呤苯并噻庚,五氟利多,培拉嗪,哌氰嗪,奋乃静,匹莫齐特,匹泮哌隆,哌泊噻嗪,丙氯拉嗪,丙嗪,异丙嗪,丙硫喷地,吡多辛,舒必利,舒托必利,丁苯那嗪,硫丙拉嗪,硫利达嗪,硫必利,替沃噻吨,三氟拉嗪,三氟丙嗪,苯海索和珠氯噻醇及其药学上可接受的盐,水合物,溶剂化物和多晶型物。
1182、在本发明的某些实施方案中,抗精神病药或其药学上可接受的盐,水合物,溶剂化物或多晶型物可以选自以下化合物:其为多巴胺能药(诸如多巴胺d1受体拮抗剂或激动剂,多巴胺d2受体拮抗剂或部分激动剂,多巴胺d3受体拮抗剂或部分激动剂,多巴胺d4受体拮抗剂),谷氨酸能药,n-甲基-d-天冬氨酸(nmda)受体正变构调节剂,甘氨酸重摄取抑制剂,谷氨酸重摄取抑制剂,促代谢的谷氨酸受体(mglurs)激动剂或正变构调节剂(pam)(例如,mglur2/3激动剂或pam),谷氨酸受体glur5正变构调节剂(pam),m1毒蕈碱样乙酰胆碱受体(machr)正变构调节剂(pam),组胺h3受体拮抗剂,α-氨基-3-羟基-5-甲基异噁唑-4-丙酸(ampa)/红藻氨酸盐受体拮抗剂,安帕金(cx-516),谷胱甘肽前药,去甲肾上腺素能剂(诸如α-2肾上腺素能受体激动剂或拮抗剂和儿茶酚-o-甲基转移酶(comt)抑制剂),血清素受体调节剂(诸如5-ht2a受体拮抗剂,5-ht1a受体部分激动剂,5-ht2c激动剂和5-ht6拮抗剂,血清素2c激动剂),胆碱能药(诸如α-7烟碱样受体激动剂或pam,α4-β2烟碱样受体激动剂,烟碱样受体的变构调节剂和乙酰胆碱酯酶抑制剂,毒蕈碱受体激动剂和拮抗剂),大麻素cb1拮抗剂,神经激肽3拮抗剂,神经降压肽激动剂,单胺氧化酶(mao)b抑制剂,pde10抑制剂,神经元一氧化氮合酶(nnos)抑制剂,神经类固醇和神经营养因子。
1183、在某些实施方案中,将如本文所述的含有α5的gabaa受体正变构调节剂和如本文所述的抗精神病药或它们的药学上可接受的盐,水合物,溶剂化物或多晶型物同时或依次或在单一制剂中或在包装在一起的分开制剂中施用。在其它的实施方案中,将含有α5的gabaa受体正变构调节剂和抗精神病药或它们的药学上可接受的盐,水合物,溶剂化物或多晶型物通过不同途径施用。如本文所用的"组合"包括通过这些制剂或施用途径中的任一种来施用。
1184、具有美金刚的药物组合物
1185、本技术的化合物或组合物可以与美金刚或其衍生物或类似物联合用于治疗有需要或处于其风险中的受试者中的与中枢神经系统(cns)障碍相关的认知损害,所述受试者包括但不限于具有年龄相关的认知损害,轻度认知损害(mci),遗忘性mci,年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症或双相型障碍,肌萎缩性侧索硬化(als)和与癌症治疗相关的认知损害或处于其风险中的受试者。
1186、美金刚,化学上也称为3,5-二甲基金刚烷-1-胺或3,5-二甲基三环[3.3.1.13,7]癸烷-1-胺,是一种具有中等亲和力的非竞争性的n-甲基-d-天冬氨酸(nmda)受体拮抗剂。美金刚的专有名称包括:和(merz),(forestlaboratories),和(lundbeck)和(unipharm)。美金刚目前在美国和世界上超过42个国家有售。其在美国被批准用于治疗中度至重度阿尔茨海默病(ad),剂量为至多28mg/天。可用于本发明的美金刚和其一些衍生物和类似物公开于美国专利号3,391,142;4,122,193;4,273,774;和5,061,703,它们都特此通过引用并入。可用于本发明的其它美金刚衍生物或类似物包括但不限于公开于以下文献中的那些化合物:美国专利申请公开us20040087658,us20050113458,us20060205822,us20090081259,us20090124659和us20100227852;欧洲专利申请公开ep2260839a2;欧洲专利ep1682109b1;和pct申请公开wo2005079779,它们都通过引用并入本文。本发明中所用的美金刚包括美金刚及其衍生物和类似物,以及其水合物,多晶型物,前药,盐和溶剂化物。本文所用的美金刚还包括包含美金刚或衍生物或类似物或其药学上可接受的盐,水合物,溶剂化物,多晶型物或前药的组合物,其中所述组合物任选地还包含至少一种另外的治疗剂(诸如可用于治疗cns障碍或其相关认知损害的治疗剂)。在某些实施方案中,适合用在本发明中的所述美金刚组合物包含美金刚和第二种治疗剂,所述第二种治疗剂是多奈哌齐(商品名aricept)。
1187、在本发明的其它实施方案中,将含有α5的gabaa受体正变构调节剂和美金刚(或美金刚衍生物/类似物)或它们的药学上可接受的盐,水合物,溶剂化物,多晶型物或前药同时或依次或在单一制剂中或在包装在一起的分开制剂中施用。在其它的实施方案中,将含有α5的gabaa受体正变构调节剂和美金刚(或美金刚衍生物/类似物)或它们的药学上可接受的盐,水合物,溶剂化物,多晶型物或前药通过不同途径施用。如本文所用的"组合"包括通过这些制剂或施用途径中的任一种来施用。
1188、具有乙酰胆碱酯酶抑制剂(ache-i)的药物组合物
1189、本技术的化合物或组合物可以与乙酰胆碱酯酶抑制剂联合用于治疗有需要或处于其风险中的受试者中的与中枢神经系统(cns)障碍相关的认知损害,所述受试者包括但不限于具有年龄相关的认知损害,轻度认知损害(mci),遗忘性mci,年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症或双相型障碍,肌萎缩性侧索硬化(als)和与癌症治疗相关的认知损害或处于其风险中的受试者。
1190、本领域普通技术人员已知的ache-i可能属于以下亚类:(i)可逆的非竞争性抑制剂或可逆的竞争性抑制剂,(ii)不可逆的,和/或(iii)拟-不可逆的抑制剂。
1191、在某些实施方案中,可用于本发明的ache-i包括在以下文献中描述的那些:pct申请wo2014039920和wo2002032412;欧洲专利号468187;481429-a;和美国专利号4,816,456;4,895,841;5,041,455;5,106,856;5,602,176;6,677,330;7,340,299;7,635,709;8,058,268;8,741,808;和8,853,219,它们都通过引用并入本文。
1192、在某些实施方案中,可依据本发明使用的典型的ache-i包括但不限于恩其明,拉多替吉,癸二胺苯酯,二乙氧膦酰硫胆碱(乙磷硫胆碱),依酚氯铵(滕西隆),他克林(盐酸他克林),解磷定(2-pam),吡啶斯的明(麦斯提龙),毒扁豆碱(丝氨酸,antilirium),abmenonium(安贝氯铵),加兰他敏(reminyl,razadyne),利凡斯的明(艾斯能,szd-ena-713),石杉碱甲,艾考哌齐,新斯的明(prostigmin,vagostigmin),安理申(多奈哌齐,e2020),山莴苣苦素,单胺吖啶类和它们的衍生物,哌啶和哌嗪衍生物,n-苄基-哌啶衍生物,哌啶基-烷酰基杂环化合物,4-(1-苄基:哌啶基)-取代的稠合喹啉衍生物和环酰胺衍生物。其它的典型的ache-i包括氨甲酸酯类和有机膦酸酯化合物类例如美曲膦酯(敌百虫)。苯并氮杂醇类(benzazepinols)诸如加兰他敏也是有用的ache-i。在某些实施方案中,适合与本技术的化合物和组合物组合使用的ache-i包括:多奈哌齐(安理申),加兰他敏(razadyne)或利凡斯的明(艾斯能)。
1193、在本发明的其它实施方案中,将含有α5的gabaa受体正变构调节剂和ache-i或它们的药学上可接受的盐,水合物,溶剂化物,多晶型物或前药同时或依次或在单一制剂中或在包装在一起的分开制剂中施用。在其它实施方案中,将含有α5的gabaa受体正变构调节剂和ache-i或它们的药学上可接受的盐,水合物,溶剂化物,多晶型物或前药通过不同途径施用。如本文所用的"组合"包括通过这些制剂或施用途径中的任一种来施用。
1194、在某些实施方案中,本文描述的化合物和组合物用于用作药物。在某些实施方案中,本发明的化合物和组合物用于在需要治疗或处于与cns障碍相关的认知损害风险中的受试者中治疗与cns障碍相关的认知损害。在某些实施方案中,所述具有认知损害的cns障碍包括但不限于,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。
1195、在某些实施方案中,本技术提供了本文描述的化合物或组合物在药物制备中的用途,所述药物用于在需要治疗或处于与cns障碍相关的认知损害风险中的受试者中治疗与cns障碍相关的认知损害。在某些实施方案中,所述具有认知损害的cns障碍包括但不限于,年龄相关的认知损害,轻度认知损害(mci),遗忘性mci(amci),年龄相关的记忆损害(aami),年龄有关的认知减退(arcd),痴呆,阿尔茨海默病(ad),前驱症状的ad,创伤后应激障碍(ptsd),精神分裂症,双相型障碍,肌萎缩性侧索硬化(als),与癌症治疗相关的认知损害,精神发育迟缓,帕金森病(pd),孤独症谱群障碍,脆性x染色体障碍,瑞特综合征,强迫行为和物质成瘾。
1196、评估认知损害的方法
1197、动物模型充当用于开发和评价与cns障碍相关的认知损害的治疗的重要资源。在动物模型中表征认知损害的特征通常扩展至人类的认知损害。因此,预期在这类动物模型中的效力会指示在人类中的效力。cns障碍的动物模型中的认知损害程度和所述cns障碍的治疗方法的效力可以使用多种认知试验进行测试并证实。
1198、放射臂迷宫(ram)行为任务是认知试验(具体地测试空间记忆)的一个例子(chappell等人,neuropharmacology 37:481-487,1998)。ram设备由例如8个等距隔开的臂组成。迷宫臂从中心平台的每个小平面伸出。食物孔位于每个臂的远侧端部处。食物用作奖赏。可以放置阻挡物以阻止进入任何臂。也可提供环绕该设备的众多额外迷宫线索。在习惯化和训练期之后,在对照或受试化合物治疗的条件下可以在ram中测试受试者的空间记忆。作为该试验的一部分,在试验之前将受试者用媒介物对照品或受试化合物的一系列剂量之一进行预处理。在每个试验开始时,将八臂迷宫的一小组臂阻挡。在该试验的最初"信息阶段"期间,允许受试者在允许进入的未阻挡的臂上得到食物。随后在信息阶段和接下来的"保留试验"之间的延迟阶段(例如,60秒延迟,15分钟延迟,1小时延迟,2小时延迟,6小时延迟,24小时延迟,或更长)将受试者从该迷宫移除,在此期间移除该迷宫上的屏障,由此允许进入所有八个臂。在延迟阶段之后,在该试验的保留试验阶段期间将受试者放回中心平台上(移除先前阻挡臂的屏障)并允许得到剩下的食物奖赏。阻挡臂的特性和配置在试验中变化。跟踪受试者在保留试验阶段形成的"错误"数。如果受试者进入已经在试验的延迟部分前从中取回食物的臂,或如果它在延迟阶段后再探访已经探访过的臂,那么错误就在试验中出现。错误数越少表明空间记忆越好。在各种受试化合物治疗方案下,可将由试验受试者做出的错误数随后用于对比受试化合物在治疗与cns障碍相关的认知损害中的效力。
1199、可用于评估受试化合物对cns障碍模型动物的认知损害的影响的另一个认知试验是莫里斯水迷宫。水迷宫是环绕着相对于迷宫的一组新奇图案的水池。用于水迷宫的训练方案可以是基于已经被证实海马依赖性的改良水迷宫任务(de hoz等人,eur.j.neurosci.,22:745-54,2005;steele和morris,hippocampus 9:118-36,1999)。训练受试者定位隐藏在水池表面下的浸没的逃逸平台。在训练试验期间,将受试者从围绕水池周围的随机开始位置放入迷宫(水池)中。开始位置在试验之间变化。如果受试者在规定的时间内未找到逃逸平台的位置,那么实验者就将受试者引导并放在平台上以"教导"它们找到平台的位置。在最后一次训练试验后的延迟阶段之后,进行不存在逃逸平台的保留试验以评估空间记忆。如通过例如由小鼠做出的在该位置花费的时间或穿过该位置的次数所测量的,受试者对(现在不存在的)逃逸平台位置的偏好水平指示更好的空间记忆,即,认知损害的治疗。然后可以将在不同治疗条件下对逃逸平台位置的偏好用于对比受试化合物在治疗与cns障碍相关的认知损害中的效力。
1200、有用于评估在人类中的认知功能的本领域已知的多种试验,例如但不限于,临床总体印象的变化量表(cibic-+量表);简短精神状态检查(mmse);神经精神调查(npi);临床痴呆等级量表(cdr);剑桥神经心理测试自动成套测试(cantab);老龄医学的桑多临床评价(scag),buschke选择联想测试(buschke和fuld,1974);口头配对相关分测试;逻辑记忆分测试;修订的韦克斯勒记忆量表的视觉复现分测试(wms-r)(wechsler,1997);本顿视觉保留试验;或精神分裂症认知功能成套测试共识版,其包括工作记忆,加工处理速度,记忆力,语言学习,视觉学习,推理和解决问题和社会认知。参见folstein等人,jpsychiatric res12:189-98,(1975);robbins等人,dementia 5:266-81,(1994);rey,l'examen cliniqueen psychologie,(1964);kluger等人,j geriatr psychiatry neurol 12:168-79,(1999);marquis等人,2002以及masur等人,1994。还参见buchanan,r.w.,keefe,r.s.e.,umbricht,d.,green,m.f.,laughren,t.,and marder,s.r.(2011)the fda-nimh-matricsguidelines for clinical trial design of cognitive-enhancing drugs:what do weknow 5years later?schizophr.bull.37,1209-1217。在人类中的认知试验的另一个实例是外显3选项强迫选择任务。在该测试中,向受试者呈现常见物体的彩色照片,该照片由以下3种类型的图像对的混合物组成:类似对,相同对和无关衬托(foils)。类似物体对的第二个称为"诱饵"。这些图像对是完全随机的,并作为系列图像单独呈现。受试者被指示做出关于所看到的物体是否是新的,旧的或类似的判断。对呈现诱饵刺激物的"类似"反应表明受试者记忆提取成功。相比之下,称诱饵刺激物为"旧的"或"新的"则表明正确的记忆提取未出现。
1201、除了评估认知表现外,还可将年龄相关的认知损害和痴呆的发展以及年龄相关的认知损害向痴呆的转化通过评价替代物在受试者脑中的变化来监测。替代物变化包括但不限于,局部脑容量的变化,穿通通路退化和通过静息态fmri(r-fmri)和氟脱氧葡萄糖正电子发射断层摄影术(fdg-pet)所见的脑功能变化。局部脑容量适用于监测年龄相关的认知损害和痴呆的发展的例子包括海马体积的减少和内嗅皮层的体积或厚度的减少。这些体积可在受试者中通过例如mri来测量。aisen等人,alzheimer's&dementia 6:239-246(2010)。已经显示穿通通路退化与年龄和降低的认知功能相关。例如,穿通通路越退化的老龄成人在海马-依赖性记忆测试中倾向于表现越差。可通过超高分辨率弥散张量成像(dti)监测在受试者中穿通通路退化。yassa等人,pnas 107:12687-12691(2010)。静息态fmri(r-fmri)涉及在静息期间成像脑,并记录在跨越功能上相关区域的暂时相关的fmri信号中大振幅自发的低频率(<0.1hz)波动。将基于种子(seed-based)的功能连接性,独立成分分析和/或信号的频域分析用于揭示在脑区之间的功能连接性,具体的是其连接性随年龄以及认知损害和/或痴呆的程度增加或减少的那些区域。fdg-pet使用fdg的摄取作为在脑中局部代谢活性的测量值。在诸如后扣带皮层,颞顶皮层和前额叶联络皮层的区域中fdg摄取的减退已经显示涉及认知减退和痴呆的程度。aisen等人,alzheimer's&dementia 6:239-246(2010),herholz等人,neuroimage 17:302-316(2002)。
1202、年龄相关的认知损害
1203、本发明提供了使用含有α5的gabaa受体正变构调节剂(即,本发明的化合物),诸如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗年龄相关的认知损害或其风险的方法和组合物。在某些实施方案中,治疗包括预防或减慢年龄相关的认知损害的发展。在某些实施方案中,治疗包括减轻,改善或减慢年龄相关的认知损害相关的一种或多种症状的发展。在某些实施方案中,年龄相关的认知损害的治疗包括减慢年龄相关的认知损害(包括但不限于mci,arcd和aami)向痴呆(例如,ad)的转化。该方法和组合物可在临床应用中用于人患者以治疗在诸如mci,arcd和aami的病症中的年龄相关的认知损害或用于其风险。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。在本发明的某些实施方案中,提供了在患有年龄相关的认知损害的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1204、在某些实施方案中,要通过本发明的方法和组合物治疗的受试者显示年龄相关的认知损害或处于这样的病损的风险中。在某些实施方案中,所述年龄相关的认知损害包括但不限于年龄相关的记忆损害(aami),轻度认知损害(mci)和年龄相关的认知减退(arcd)。
1205、动物模型充当用于开发和评价这样的年龄相关的认知损害的治疗的重要资源。在动物模型中表征年龄相关的认知损害的特征通常扩展至人类中的年龄相关的认知损害。因此,预见到在这样的动物模型中的效力指示在人类中的效力。
1206、年龄相关的认知损害的多种动物模型是本领域已知的。例如,大量的行为特征已鉴定了在远交系老龄long-evans大鼠中认知损害的自然发生形式(charles riverlaboratories;gallagher等人,behav.neurosci.107:618-626,(1993))。在用莫里斯水迷宫(mwm)进行的行为评价中,大鼠学习并记忆由该迷宫周围的空间线索配置引导的逃逸平台的位置。使用动物在搜寻所述逃逸平台的位置中空间偏好的测量值,在探查试验中测试表现的认知基础。在该研究群体中老龄大鼠游至可见的平台并无困难,但是当将该平台伪装,需要利用空间信息时,就检测到了年龄依赖性损害。在远交long-evans系中的个别老龄大鼠的表现有很大不同。例如,部分那些大鼠与年轻成年大鼠有同样表现。但是,约40-50%超出了年轻大鼠表现的范围。在老龄大鼠中的这种可变性反映了可靠的个体差异。因此,在老龄群体中一些动物是认知受损的,并称为老龄受损(ai),而其它动物是未受损的,并称为老龄未受损(au)。参见,例如,colombo等人,proc.natl.acad.sci.94:14195-14199,(1997);gallagher和burwell,neurobiol.aging 10:691-708,(1989);gallagher等人,behav.neurosci.107:618-626,(1993);rapp和gallagher,proc.natl.acad.sci.93:9926-9930,(1996);nicolle等人,neuroscience 74:741-756,(1996);nicolle等人,j.neurosci.19:9604-9610,(1999);国际专利公开文本wo2007/019312和国际专利公开文本wo2004/048551。年龄相关的认知损害的这类动物模型可用于测试本发明的方法和组合物在治疗年龄相关的认知损害中的有效性。
1207、使用多种认知试验(包括如上所讨论的莫里斯水迷宫和放射臂迷宫)可以评估本发明的方法和组合物在治疗年龄相关的认知损害中的效力。
1208、痴呆
1209、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗痴呆的方法和组合物。在某些实施方案中,治疗包括预防或减慢痴呆的发展。在某些实施方案中,治疗包括减轻,改善或减慢与痴呆相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害。在本发明的某些实施方案中,提供了在患有痴呆的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在某些实施方案中,所述痴呆是阿尔茨海默病(ad),血管性痴呆,痴呆伴露易小体或额颞叶痴呆。该方法和组合物可在临床应用中用于人患者以治疗痴呆。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。
1210、动物模型充当用于开发和评价痴呆的治疗的重要资源。在动物模型中表征痴呆的特征通常扩展至人类中的痴呆。因此,预见到在这样的动物模型中的效力指示在人类中的效力。痴呆的各种动物模型是本领域已知的,诸如pdapp,tg2576,app23,tgcrnd8,j20,hps2tg和app+ps1转基因小鼠。sankaranarayanan,curr.top.medicinal chem.6:609-627,2006;kobayashi等人,genes brain behav.4:173-196.2005;ashe和zahns,neuron.66:631-45,2010。痴呆的这类动物模型可用于测定本发明的方法和组合物在治疗痴呆中的有效性。
1211、使用如本文所讨论的本领域已知的多种认知试验,可以在痴呆的动物模型以及患痴呆的人受试者中评估本发明的方法和组合物在治疗痴呆或与痴呆相关的认知损害中的效力。
1212、创伤后应激障碍
1213、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗创伤后应激障碍(ptsd)的方法和组合物。在某些实施方案中,治疗包括预防或减慢ptsd的发展。在某些实施方案中,治疗包括减轻,改善或减慢与ptsd相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害。在本发明的某些实施方案中,提供了在患有ptsd的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。该方法和组合物可在临床应用中用于人患者以治疗ptsd。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。
1214、患ptsd的患者(和暴露于较小程度创伤,未患ptsd的患者)有更小的海马体积(woon等人,prog.neuro-psychopharm.&biological psych.34,1181-1188;wang等人,arch.gen.psychiatry 67:296-303,2010)。ptsd也与受损认知表现相关。患ptsd的老龄个体相对于对照患者,认知表现有更大减退(yehuda等人,bio.psych.60:714-721,2006)并具有发展痴呆的更大可能性(yaffe等人,arch.gen.psych.678:608-613,2010)。
1215、动物模型充当用于开发和评价ptsd的治疗的重要资源。在动物模型中表征ptsd的特征通常扩展至人类中的ptsd。因此,预见到在这样的动物模型中的效力指示在人类中的效力。ptsd的各种动物模型是本领域已知的。
1216、ptsd的一个大鼠模型是时间依赖性致敏作用(time-dependent sensitization)(tds)模型。tds涉及使动物暴露于强烈应激事件,之后进行先前应激的处境提醒。下列是tds的例子。将大鼠置于制动器中,然后置于泳池中并使其游泳一段时间,例如,20分钟。随后,立即将每个大鼠暴露于气态麻醉剂中直到意识丧失,并最终干燥。使动物不受打扰持续许多天,例如,一周。然后将大鼠暴露于包括初始应激源的"再次应激"期,例如,在泳池中游泳期(liberzon等人,psychoneuroendocrinology22:443-453,1997;harvery等人,psychopharmacology 175:494-502,2004)。tds引起大鼠增强的听觉惊恐反应(asr),其可与夸张的听觉惊恐(为ptsd的显著症状)相比(khan和liberzon,psychopharmacology172:225-229,2004)。ptsd的这类动物模型可用于测试本发明的方法和组合物在治疗ptsd中的有效性。
1217、使用如本文所讨论的本领域已知的多种认知试验,也可以在ptsd的动物模型以及患ptsd的人受试者中评估本发明的方法和组合物在治疗ptsd或与ptsd相关的认知损害中的效力。
1218、精神分裂症和双相型障碍
1219、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗精神分裂症或双相型障碍(特别是躁狂症)的方法和组合物。在某些实施方案中,治疗包括预防或减慢精神分裂症或双相型障碍(特别是躁狂症)的发展。精神分裂症的特征在于广谱精神病理学情况,包括阳性症状,诸如异常或扭曲的心理表征(例如幻觉,妄想),或用于多巴胺失调-相关的症状(例如,高多巴胺能反应(hyperdopaminergic responses),高多巴胺能行为反应,多巴胺能机能亢进或高活动性(hyperlocomotor activity)或精神病),阴性症状,其特征在于动机减少和适应性目标定向作用(例如兴趣缺失,情感冷淡,缺乏情感反应)和认知损害。在某些实施方案中,治疗包括减轻,改善或减慢与精神分裂症相关的一个或多个阳性和/或阴性症状以及认知损害的发展。此外,存在许多其它精神病,诸如典型精神分裂症(schizotypical disorder)和情感性分裂症,其它急性和慢性精神病和双相型障碍(特别是躁狂症),它们具有与精神分裂症重叠的复合症状。在某些实施方案中,治疗包括减轻,改善或减慢与双相型障碍(特别是躁狂症)相关的一个或多个症状的发展以及认知损害。在本发明的某些实施方案中,提供了在患有精神分裂症或双相型障碍的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。该方法和组合物可在临床应用中用于人患者以治疗精神分裂症或双相型障碍(特别是躁狂症)。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。
1220、认知损害也与精神分裂症相关。它们先于精神病的发作,并且在未受影响的亲属(relatives)中存在。与精神分裂症相关的认知损害构成功能后果的良好预报器,并且是该障碍的核心特征。精神分裂症中的认知特征反映在额皮层和海马回路中的功能障碍。患精神分裂症的患者也存在诸如海马体积减少,神经元尺寸减少和功能障碍性活动过强的海马病状。也已在精神分裂症的患者中记录到这些脑区中兴奋和抑制的失衡,其暗示药物靶向抑制的机制可能是有疗效的。参见,例如,guidotti等人,psychopharmacology 180:191-205,2005;zierhut,psych.res.neuroimag.183:187-194,2010;wood等人,neuroimage 52:62-63,2010;vinkers等人,expert opin.investig.drugs 19:1217-1233,2009;young等人,pharmacol.ther.122:150-202,2009。
1221、动物模型充当用于开发和评价精神分裂症的治疗的重要资源。在动物模型中表征精神分裂症的特征通常扩展至人类中的精神分裂症。因此,预见到在这样的动物模型中的效力指示在人类中的效力。精神分裂症的各种动物模型是本领域已知的。
1222、精神分裂症的一个动物模型是用蛋氨酸长久处理。蛋氨酸处理的小鼠显示在额皮层和海马中缺乏gad67的表达,类似于在精神分裂症患者死后脑中所报道的情况。它们也显示惊恐和社交缺陷的前冲动抑制(tremonlizzo等人,pnas,99:17095-17100,2002)。精神分裂症的另一个动物模型为在大鼠中用甲基氧化偶氮甲醇乙酸盐(mam)处理。在妊娠第17天向怀孕雌性大鼠施用mam(20mg/kg,腹膜内)。mam-处理概括了在后代中精神分裂症-样表型的病理发展过程,包括解剖学变化,行为缺陷和改变的神经元信息处理。更具体的是,mam处理的大鼠显示小白蛋白-阳性gaba能中间神经元在前额皮层和海马部分中密度降低。在行为测试中,mam处理的大鼠显示减小的潜伏抑制。潜伏抑制是一种行为现象,其中减少了对刺激物的学习,对于该刺激物存在具有任何结果的先前暴露。忽视先前良性刺激和减少与这类刺激相关的信息的倾向被认为防止了感觉超负荷。潜伏抑制低表明精神病。潜伏抑制可在大鼠中以下列方式测试。将大鼠分成两组。将一组在多次试验中预暴露于一个声调下。另一组不呈现声调。然后将两组暴露于听觉恐惧条件反射程序中,其中相同的声调与伤害性刺激,诸如电击足同时呈现。随后,向两组呈现该声调,并在声调呈现期间监测大鼠运动行为的变化。在恐惧条件反射之后,大鼠通过极力减少运动行为对声调呈现做出反应。但是,在条件反射期之前已暴露于该声调中的组显示强烈的潜伏抑制:响应声调呈现的运动行为抑制减少了。mam处理的大鼠,相比之下显示受损潜伏抑制。也就是,在恐惧条件反射程序之前暴露于该声调,在抑制恐惧条件反射中并不具有显著作用。(参见lodge等人,j.neurosci.,29:2344-2354,2009)。精神分裂症的这类动物模型可用于测试本发明的方法和组合物在治疗精神分裂症或双相型障碍(特别是躁狂症)中的有效性。
1223、mam-处理的大鼠对低剂量右苯丙胺施用展示出显著增强的运动响应(或异常运动行为)。mam-处理的大鼠还展示出明显巨大数量的自发激发腹侧盖多巴胺(da)神经元。认为这些结果是过度海马活动的结果,因为在mam-处理的大鼠中,腹侧海马回(vhipp)失活(例如因vhipp内施用钠通道阻滞剂河豚毒素(ttx)给mam大鼠)完全逆转了升高的da神经元群体活动并且还纠正了增强的苯丙胺诱导的运动行为。认为海马功能障碍与da系统的过度响应率的相关性以对mam-处理的动物中的苯丙胺响应增强和精神分裂症患者中的精神病为基础。参见lodge d.j.等人,neurobiology of disease(2007),27(42),11424-11430。mam-处理的大鼠在上述研究中的应用可以适用于测定本发明的方法和组合物在治疗精神分裂症或双相型障碍(特别是躁狂症)中的有效性。例如,可以使用mam-处理的动物评价本发明的方法和组合物对中枢海马(vhipp)调节,升高的da神经元群体活动和对mam-处理的动物中苯丙胺的活动过强响应的影响。
1224、在mam-处理的大鼠中,海马(hpc)功能障碍导致多巴胺系统活动过度。测试对gabaa受体α5亚基具有选择性的苯并二氮杂环庚三烯-正变构调节剂(pam)sh-053-2'f-r-ch3对海马(hpc)输出量的影响。还检查了sh-053-2'f-r-ch3对苯丙胺在mam-处理动物中的活动过强的运动响应的影响。当全身施用和直接输注入腹侧hpc时,α5gabaar pam将在mam大鼠的腹侧盖区域(vta)中的自发活动的da神经元数量降至在盐水处理的大鼠(对照组)中观察到的水平。此外,盐水处理和mam-处理的动物中的hpc神经元在α5gabaar pam治疗后显示减少的皮质诱发反应。此外,对在mam-处理大鼠中观察到的对苯丙胺的运动响应增加在α5gabaar pam治疗后减少。参见gill k.m等人neuropsychopharmacology(2011),1-9。mam-处理的大鼠在上述研究中的应用可以适用于本发明测定本发明的方法和组合物在治疗精神分裂症或双相型障碍(特别是躁狂症)中的有效性。例如,可以使用mam-处理的动物评价本发明的方法和组合物对海马(hpc)输出量和对苯丙胺在mam-处理的动物中的活动过强响应的影响。
1225、在胚胎第15天(e15)时施用mam给妊娠大鼠严重地影响了子代中空间记忆或学习在八臂放射迷宫上对4个物品的空间定位的能力。此外,胚胎第17天(e17)mam-处理的大鼠能够在训练的起始阶段达到对照大鼠的行为水平,但在插入30min延迟时不能加工和恢复空间信息,表明工作记忆显著损害。参见,gourevitch r.等人,(2004),behav.pharmacol,15,287-292。这类精神分裂症的动物模型可以用于测定本发明的方法和组合物在治疗精神分裂症或双相型障碍(特别是躁狂症)中的有效性。
1226、阿扑吗啡诱导的小鼠中的攀爬(aic)和刻板(ais)是用于本发明的另一种动物模型。以期望的剂量水平(例如通过腹膜内施用)给小鼠施用试剂。随后,诸如在30分钟后,用阿扑吗啡(例如1mg/kg皮下)攻击实验小鼠。阿扑吗啡注射后5分钟,对每只动物给由阿扑吗啡诱导的鼻吸气-舔-撕咬综合征(刻板行为)和攀爬行为评分并且记录。在30min测试期间每隔5min重复读取。将30min测试期间每只动物的每种综合征(刻板行为和攀爬)的得分汇总。条件是效果达到至少50%抑制,且使用非线性最小二乘法计算与反向预测来计算id50值(95%置信区间)。可以将平均攀爬和刻板得分表示为在接受阿扑吗啡的媒介物治疗(例如盐水治疗的)的小鼠中观察到的对照值的百分比。参见grauer s.m.等人,psychopharmacology(2009)204,37-48。这种小鼠模型可以用于测定本发明的方法和组合物在治疗精神分裂症或双相型障碍(特别是躁狂症)中的有效性。
1227、在精神分裂症的另一个充分确立的临床前模型中,长期暴露于氯胺酮(非竞争性n-甲基-d-天冬氨酸(nmda)受体拮抗剂)的大鼠产生了阳性和阴性精神病症状和认知损害。在青春期(2个月大),long-evans雄性大鼠经腹膜内注射氯胺酮(30mg/kg,每日两次)达两周。当大鼠达到成年时(约4-5个月大),对它们进行行为测试,目的是测试对氯胺酮暴露的行为症状和为了测试用于缓解这些症状的治疗的效力。参见,例如,enomoto等人progressin neuro-psychopharmacology&biological psychiatry 33(2009)668-675。
1228、使用如本文所讨论的本领域已知的多种认知试验,也可以在精神分裂症或双相型障碍(特别是躁狂症)的动物模型以及患精神分裂症的人受试者中评估本发明的方法和组合物在治疗精神分裂症或与其相关的认知损害中的效力。
1229、肌萎缩性侧索硬化(als)
1230、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗als的方法和组合物。在某些实施方案中,治疗包括预防或减慢als的发展。在某些实施方案中,治疗包括减轻,改善或减慢与als相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害。在本发明的某些实施方案中,提供了在患有als的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。该方法和组合物可在临床应用中用于人患者以治疗als。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。
1231、除了运动神经元变性外,als表征为在内嗅皮层和海马中神经元变性,记忆缺陷以及在诸如皮层的不同脑区中神经元兴奋性过高。
1232、使用如本文所讨论的本领域已知的多种认知试验,也可以在als的动物模型以及患als的人受试者中评估本发明的方法和组合物在治疗als或与als相关的认知损害中的效力。
1233、与癌症治疗相关的认知损害
1234、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗与癌症治疗相关的认知损害的方法和组合物。在某些实施方案中,治疗包括预防或减慢与癌症治疗相关的认知损害的发展。在某些实施方案中,治疗包括减轻,改善或减慢与癌症治疗相关的认知损害相关的一种或多种症状的发展。在本发明的某些实施方案中,提供了在患有与癌症治疗相关的认知损害的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。该方法和组合物可在临床应用中用于人患者以治疗与癌症治疗相关的认知损害。如本文所述,用于所述方法的组合物的剂量和剂量间隔是在那些应用中安全的和有效的。
1235、用于癌症治疗的治疗(包括化学治疗,辐射或其组合)可引起患者在如记忆,学习和注意力的这类功能中的认知损害。对癌症治疗的脑的细胞毒性和其它不良副作用以这种形式的认知损害为基础,该认知障碍可持续十年。(dietrich等人,oncologist 13:1285-95,2008;soussain等人,lancet 374:1639-51,2009)。
1236、癌症治疗之后的认知损害反映了正常认知所必需的额皮层和海马回路功能障碍。在动物模型中,暴露于化学治疗或辐射对认知特异性依赖于这些脑系统特别是海马的测试表现有不利的影响(kim等人,j.radiat.res.49:517-526,2008;yang等人,neurobiol.learning and mem.93:487-494,2010)。因此,靶向这些皮层和海马系统的药物在接受癌症治疗患者中可以是神经保护的,并且在治疗认知损害的症状中是有效的,可持续超过用作癌症治疗的干预。
1237、动物模型充当用于开发和评价与癌症治疗相关的认知损害的治疗的重要资源。在动物模型中表征与癌症治疗相关的认知损害的特征通常扩展至人类中的与癌症治疗相关的认知损害。因此,预见到在这样的动物模型中的效力指示在人类中的效力。与癌症治疗相关的认知损害的各种动物模型是本领域已知的。
1238、与癌症治疗相关的认知损害的动物模型的例子包括用诸如环磷酰胺(cyp)的抗肿瘤剂或用辐射(例如,60coγ射线)治疗动物。(kim等人,j.radiat.res.49:517-526,2008;yang等人,neurobiol.learning和mem.93:487-494,2010)。然后可用认知试验来测试与癌症治疗相关的认知损害的动物模型的认知功能,以测试本发明的方法和组合物在治疗与癌症治疗相关的认知损害中的有效性。使用如本文所讨论的本领域已知的多种认知试验,评估本发明的方法和组合物在治疗与癌症治疗相关的认知损害以及患与癌症治疗相关的认知损害的人受试者中的效力。
1239、帕金森病(pd)
1240、帕金森病(pd)是特征在于随意运动减少的神经障碍。患病的患者与正常个体相比具有运动活动减少和较为缓慢的随意运动。该患者具有特征性"面具"脸,行走同时的匆忙倾向,弯曲的体姿和肌肉泛发性虚弱。存在典型的"铅管样"强直的被动运动。该病的另一个重要特征在于在休息时的四肢震颤和运动过程中的减少。
1241、帕金森病(其病因尚不了解)属于一组最常见的运动障碍,称作帕金森综合征,其影响每1000人中的约1个人。在帕金森综合征名称下分组的这些另外的障碍可以因病毒感染,梅毒,动脉硬化和创伤和暴露于毒性化学药品和麻醉品导致。但是,认为突触稳定性的不适当缺失可以导致神经元回路破坏和脑疾病。无论是遗传,药物使用,衰老过程,病毒感染,还是其它各种原因的结果,认为神经元通讯中的功能障碍是许多神经病的主要原因,诸如pd(myrrhe van spronsen和casper c.hoogenraad,curr.neurol.neurosci.rep.2010,10,207-214)。
1242、与疾病的原因无关,主要的病理性特征是基底神经节,尤其是黑质中的多巴胺能细胞变性。由于黑质中包含多巴胺的神经元过早死亡,所以基底神经节的最大结构纹状体已经减少了从黑质的输入,导致多巴胺释放减少。对主要病理学的理解导致引入了第一种成功的治疗方法,其可以缓解帕金森病。实际上,所有针对该病治疗的方法均基于多巴胺替代。用于治疗的药物可以在交叉通过血脑屏障后被转化成多巴胺,或它们可以加强多巴胺的合成并且减少其分解。不幸的是,主要的病理学情况黑质中细胞的变性未得以解除。该病持续进展并且在一定时间期限后频繁,多巴胺替代疗法失去其有效性。
1243、本发明提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗pd的方法和组合物。在某些实施方案中,治疗包括预防或减慢pd的发展。在某些实施方案中,治疗包括减轻,改善或减慢与pd相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害。例如,本发明的方法和组合物可以用于改善帕金森病的活动/认知损害症状。此外,本公开文本的方法和组合物可以用于治疗帕金森病的记忆损害症状。在本发明的某些实施方案中,提供了在患有pd的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1244、存在许多pd的动物模型。pd的示例性的动物模型包括利血平模型,甲基苯丙胺模型,6-羟基多巴胺(6-ohda)模型,1-甲基-4-苯基-1,2,3,6-四氢吡啶(mptp)模型,百草枯(pq)-代森锰模型,鱼藤酮模型,3-硝基酪氨酸模型和使用转基因小鼠的遗传学模型。转基因模型包括超表达α-突触核蛋白,表达α-突触核蛋白的人突变型的小鼠或表达lrkk2突变的小鼠。参见ranjita b.等人的这些模型的综述(ranjita b.等人,bioessays2002,24,308-318)。有关这些动物模型的另外的信息易于得自jackson laboratories(还参见http://research.jax.org/grs/parkinsons.html)以及公开这些经验证的模型应用的大量出版物。
1245、使用如本文所讨论的本领域已知的多种认知试验,可以在任意上述pd动物模型以及在具有pd的人受试者中评估本发明的方法和组合物在治疗pd或与pd相关的认知损害中的效力。
1246、孤独症
1247、孤独症是特征在于三种核心行为等级功能障碍的神经发育障碍:重复行为,社交缺乏和认知缺陷。重复行为领域包括强迫行为,对目标罕见依恋,顽固性的墨守成规或礼仪行为和反复性的活动作态,诸如刻板和自我激动行为。社交缺陷等级包括社会交流缺陷,缺乏目光接触,从事会话的能力减弱和每日社交机能受损。认知缺陷可以包括语言异常。孤独性障碍是致残性神经障碍,其影响数以千计的美国人并且包括大量亚型,具有不同的推定原因且几乎没有文献记载改善性的治疗。孤独性谱群疾病可以在出生时存在或可能随后发作,例如在2或3岁时。没有明确的孤独症的生物学标记。该障碍的诊断由考虑儿童匹配行为综合征的程度进行,所述行为综合征的特征在于畅谈能力差,社交和认知能力怪癖和不适合的行为模式。神经元通讯功能障碍被视为孤独症的主要原因之一(myrrhe van spronsen和casper c.hoogenraad,curr.neurol.neurosci.rep.2010,10,207-214)。最新研究已经显示孤独症谱群疾病(asd)中存在gabaaα5缺陷,并支持在该疾病中gaba系统的进一步研究(mendez ma,等人neuropharmacology.2013,68:195-201)。
1248、本发明还提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗孤独症的方法和组合物。在某些实施方案中,治疗包括预防或减慢孤独症的发展。在某些实施方案中,治疗包括减轻,改善或减慢与孤独症相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害或认知缺陷。例如,本发明的方法和组合物可以用于改善孤独症的活动/认知缺陷症状。在本发明的某些实施方案中,提供了在患有孤独症的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1249、使用rodier等人(rodier,p.m.等人reprod.toxicol.1997,11,417-422)建立的体外电生理技术的孤独症丙戊酸(vpa)的大鼠模型是基于最充分建立的创伤的孤独症动物模型之一,并且基于如下观察结果:在胚胎发育限制的时间窗期间,在上世纪60年代用vpa治疗的孕妇生育孤独症儿童的风险远高于正常人群。暴露于vpa的妊娠大鼠的子代显示几个典型孤独症的解剖和行为症状,诸如小脑普肯耶神经元数量减少,社会相互作用削弱,重复行为和其它孤独症症状,包括恐惧记忆加工处理增强。参见rinaldi t.等人frontiers inneural circuits,2008,2,1-7。另一个小鼠模型,btbr t+tf/j(btbr)小鼠,是具有与孤独症的三种诊断的行为症状(不正常的社会交往,受损的沟通和重复的行为)相关的稳健的行为表型的被确立的模型,其被用于探查mglur5受体的选择性负变构调节剂grn-529的效力。参见例如,silverman j.l.等人sci transl.med.2012,4,131。使用如本文所讨论的本领域已知的多种认知试验,可以在vpa-治疗的孤独症大鼠模型或btbr t+tf/j(btbr)小鼠模型以及具有孤独症的人受试者中评估本发明的方法和组合物在治疗孤独症或与孤独症相关的认知缺陷中的效力。
1250、精神发育迟缓
1251、精神发育迟缓是一种特征在于显著受损的认知功能和适应行为缺陷的泛发性障碍。精神发育迟缓通常被定义为小于70的智商(iq)得分。在许多潜在的原因中,精神发育迟缓是先天性原因。神经元通讯功能障碍也被视为精神发育迟缓的潜在原因之一(myrrhevan spronsen和casper c.hoogenraad,curr.neurol.neurosci.rep.2010,10,207-214)。
1252、在某些情况下,精神发育迟缓包括但不限于唐氏综合征,腭-心-面综合征,胎儿酒精综合征,脆性x染色体综合征,克兰费尔特综合征,神经纤维瘤病,先天性甲状腺功能减退症,威廉斯综合征,苯丙酮尿(pku),史-伦-奥三氏综合征,帕-魏二氏综合征,phelan-mcdermid综合征,mowat-wilson综合征,纤毛类疾病,lowe综合征和siderium型x-连锁智力障碍。唐氏综合征是一种障碍,其包括初生缺陷包括一定程度的精神发育迟缓,特征性面部特征和通常的心脏缺陷,感染增加,与视觉和听觉相关的问题和其它健康问题的组合。脆性x染色体综合征是遗传性精神发育迟缓的普遍形式,其发生频率为4,000个男性中有1个和8,000个女性中有1个。这种综合征的特征还在于发展迟滞,活动过度,注意缺陷障碍和孤独样行为。对于脆性x染色体综合征没有有效的治疗方法。
1253、本发明预见到轻度精神发育迟缓,中度精神发育迟缓,严重精神发育迟缓,深度精神发育迟缓和未详细说明严重程度的精神发育迟缓的治疗方法。这类精神发育迟缓可以,但不一定与染色体改变(例如唐氏综合征归因于三体性21),遗传性,妊娠和围产期问题和其它严重精神障碍相关。本发明提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗精神发育迟缓的方法和组合物。在某些实施方案中,治疗包括预防或减慢精神发育迟缓的发展。在某些实施方案中,治疗包括减轻,改善或减慢与精神发育迟缓相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知缺陷/认知损害。例如,本公开内容的方法和组合物可以用于改善精神发育迟缓的活动/认知损害症状。在本发明的某些实施方案中,提供了在患有精神发育迟缓的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1254、已经为精神发育迟缓开发了几种动物模型。例如,已经为脆性x染色体综合征开发了敲除小鼠模型。脆性x染色体综合征是因不存在fmr1蛋白fmrp导致的精神发育迟缓的常见形式。已经鉴定了fmrp的两种同源物fxr1p和fxr2p。fxr2p显示在脑和睾丸中高度表达,如fmrp。认为fxr2和fmr1敲除小鼠和fmr1/fxr2双敲除小鼠是精神发育迟缓诸如脆性x染色体综合征的有用的模型。参见bontekoe c.j.m.等人hum.mol.genet.2002,11(5):487-498。使用如本文所讨论的本领域已知的多种认知试验,可以在这些小鼠模型和其它为精神发育迟缓开发的动物模型以及具有精神发育迟缓的人受试者中评估本发明的方法和组合物在治疗精神发育迟缓或与精神发育迟缓相关的认知缺陷/损害中的效力。
1255、强迫行为(强迫性障碍)
1256、强迫性障碍("ocd")是一种精神病症,其最常见的特征在于导致个体感觉被驱使执行的强迫行为和心理活动(强迫行为)的插入性的,反复性的不需要的思维(强迫观念)。当前的流行病学数据表明ocd是美国第四位最常见的精神障碍。一些研究表明ocd的发病率在1%-3%之间,尽管临床公认的ocd的发病率低得多,从而提示,许多具有该障碍的个体可能未被诊断。具有ocd的患者通常由心理学家,精神病专家或精神分析学家根据《精神病诊断与统计手册》(diagnostic and statistical manual of mental disorders,第4版修订版(dsm-iv-tr)(2000)诊断标准(包括强迫观念行为和强迫行为的特征)来诊断。强迫观念的特征包括:(1)作为插入而经历并且导致显著焦虑或痛苦的反复和持续性的思维,冲动或想象;(2)思维,冲动或想象不简单地是对有关现时问题的过度担忧;和(3)人尝试忽略或抑制这类思维,冲动或想象,或用一些其它思维或行动中和它们。人认为强迫观念的思维,冲动或想象是他/她自身思想的产物且不基于现实性。强迫行为的特征包括:(1)人感觉被驱使响应于强迫观念或根据必须严格遵守的规则而做出的重复性行为或心理活动;(2)行为或心理活动的目的在于预防或减少苦恼或预防一些令人畏惧的事件或情况;然而,这些行为或心理活动实际上与问题无关,或它们是过度的。
1257、具有ocd的个体典型地完成任务(或强迫行为)以寻求从与强迫观念相关的焦虑中解除。借助于强迫观念思维或脱离它们通常进行反复性的行为,诸如洗手,计数,检查或清洁。然而,完成这些"仪式"仅提供短暂的解脱。具有ocd的人还可以使用其它精神障碍谱诊断,诸如广泛性焦虑症,神经性厌食,惊恐发作或精神分裂症。
1258、神经元通讯中的功能障碍被视为强迫障碍的根本原因之一(myrrhe vanspronsen和casper c.hoogenraad,curr.neurol.neurosci.rep.2010,10,207-214)。研究提示,ocd与称作血清素的神经递质异常水平相关。ocd的一线治疗由行为疗法,认知疗法和药物组成。治疗用药包括血清素再摄取抑制剂(sri),诸如帕罗西汀(seroxattm,xetanortm,paromercktm,rexetintm),舍曲林(stimulotontm),氟西汀(bioxetintm),艾司西酞普兰和氟伏沙明以及三环抗抑郁药,特别是氯米帕明苯并二氮杂环庚三烯类也用在治疗中。然而,高达40-60%的患者没有对sri疗法做出适当响应,且甚至更大比例的患者没有经历其症状的完全缓解。
1259、本发明提供了使用含有α5的gabaa受体激动剂(例如,含有α5的gabaa受体正变构调节剂),例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗ocd的方法和组合物。在某些实施方案中,治疗包括预防或减慢ocd的发展。在某些实施方案中,治疗包括减轻,改善或减慢与ocd相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害或认知缺陷。例如,本公开内容的方法和组合物可以用于治疗ocd中的认知缺陷和/或改善具有ocd的患者的认知功能。在本发明的某些实施方案中,提供了在患有ocd的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1260、已经为ocd开发了喹吡罗致敏的大鼠模型。将喹吡罗致敏的大鼠的强迫检查行为进行中断,其为ocd强迫行为的特有属性。此外,使用强迫性障碍的程序-诱导的烦渴(sip)啮齿动物模型来评价新的5-ht2c受体激动剂way-163909的效力。参见,例如rosenzweig-lipson s.等人psychopharmacology(berl)2007,192,159-70。使用如本文所讨论的本领域已知的多种认知试验,可以在为ocd开发的以上动物模型和其它动物模型中以及具有ocd的人受试者中评估本发明的方法和组合物在治疗ocd或与ocd相关的认知损害或认知缺陷中的效力。
1261、物质成瘾
1262、物质成瘾(例如药物成瘾,酒精成瘾)是一种精神障碍。物质成瘾不是在接触滥用物质后立即被触发。更确切地,它涉及在数小时至数日至数个月范围的不同时间内发生的多个复杂的神经改变(kauer j.a.nat.rev.neurosci.2007,8,844-858)。物质成瘾的路径一般开始于自愿使用一种或多种管制物质,诸如麻醉品,巴比妥酸盐类,甲基苯丙胺,酒精,烟碱和多种其它这样的管制物质中的任一种。随时间的推移,由于管制物质的延长使用,自愿避开管制物质的能力因延长使用对脑功能的影响和由此对行为的影响而受损。这样,物质成瘾一般特征在于强迫的物质渴求,寻找和使用,甚至在面对消极后果时仍然持续。渴求可以代表患者的潜在神经生物学改变,如果需要恢复,则可能必须以有意义的方式解决所述改变。在许多情况下,物质成瘾还特征在于戒断症状,所述戒断症状对于一些物质(例如酒精,巴比妥酸盐类)而言是威胁生命的,而在其它情况下可以导致显著的病态(其可以包括恶心,呕吐,发热,眩晕和大量出汗),痛苦和降低的得到恢复的能力。例如,酒精中毒(也称作酒精依赖性)是一种这样的物质成瘾。酒精中毒的主要特征在于4种症状,包括渴求,失控,身体依赖性和耐受性。这些症状还可以表征对其它管制物质的物质成瘾。对酒精以及其它管制物质的渴求经常与对食物或水的需求一样强烈。因此,尽管存在严重的家庭,健康和/或法律分歧,但是可以继续饮用酒精。
1263、探究滥用酒精,中枢兴奋药和阿片类物质对中枢神经系统(cns)的影响的最近工作已经证实了与精神卫生相关的多种不良作用,包括物质诱导的认知损害。参见nybergf.cognitive impairments in drug addicts,第9章。在几个实验室和诊所中观察到这些药物导致脑功能实质损害。在滥用药物对脑的有害作用中有促成加速退化的那些。在近年期间已经引起特别关注的一项观察结果是,长期药物使用者展示出与执行和记忆功能相关的脑区域中的显著损害。由成瘾药物诸如酒精,中枢兴奋药和阿片类物质导致的显著的神经适应涉及海马的亚粒状区(sgz)中的神经发生减少。实际上,已经提出在sgz中减少的成年神经发生可以以促成复发和维持性的成瘾行为的方式改变海马功能。它还产生了以下可能性:减少的神经发生可以促成由这些滥用药物引起的认知缺陷。
1264、本发明提供了使用含有α5的gabaa受体正变构调节剂,例如选自如本文所述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合中的一种,治疗物质成瘾的方法和组合物。在某些实施方案中,治疗包括预防或减慢物质成瘾的发展。在某些实施方案中,治疗包括减轻,改善或减慢与物质成瘾相关的一种或多种症状的发展。在某些实施方案中,待治疗的症状是认知损害。例如,本公开内容的方法和组合物可以用于在具有物质成瘾的患者中治疗认知损害和/或改善认知功能。在本发明的某些实施方案中,提供了在具有物质成瘾的受试者中保留或改善认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。
1265、已经开发了几种动物模型以研究物质成瘾。例如,开发了遗传上选择的marchigian sardinian偏好酒精的(msp)大鼠模型以研究酒精中毒的神经生物学。参见ciccocioppo r.等人substance addiction biology 2006,11,339-355。使用如本文所讨论的本领域已知的多种认知试验,可以在物质成瘾的动物模型和具有物质成瘾的人受试者中评估本发明的方法和组合物在治疗物质成瘾或与物质成瘾相关的认知损害中的效力。
1266、脑癌
1267、脑癌是在脑组织中的异常细胞生长,通常涉及恶性脑肿瘤的生长。脑肿瘤生长且压迫附近的脑区域,其能够使得该部分脑停止发挥其应有的功能。脑癌很少扩展入脑以外的其它组织。基于癌细胞在显微镜下所展现的异常程度的肿瘤等级可以用来区别缓慢与快速生长肿瘤之间的差异。脑肿瘤根据肿瘤显得自其发源的细胞种类来分类。弥漫性、纤维性星形细胞瘤是成人原发脑肿瘤的最常见形式。这些肿瘤在组织病理学上分为三个恶性等级:世界卫生组织(who)ii级星形细胞瘤,who iii级间变性星形细胞瘤和who iv级多形性成胶质细胞瘤(gbm)。who ii级星形细胞瘤是弥漫性星形细胞瘤谱中最不活跃的。星形细胞瘤显示渗入周围脑的显著倾向,使得局部控制的治疗尝试变得困难。这些侵袭能力在低级以及高级肿瘤中常常是明显的。
1268、多形性成胶质细胞瘤是星形细胞瘤的最恶性阶段,大多数患者的存活时间小于2年。在组织学上,这些肿瘤的特征是致密的细胞构成,高增殖指数,内皮增殖和局灶性坏死。这些损伤的高度增殖性可能来自多种致有丝分裂效果。gbm的标志之一内皮增殖。在gbm中发现多种血管生成生长因子及其受体。
1269、存在星形细胞瘤的生物学亚型,其可以反映在这些肿瘤中观察到的临床多相性。这些亚型包括脑干胶质瘤(其是儿科弥漫性形式),常常在恶性过程之后的纤维性星形细胞瘤。脑干gbm与影响更年轻患者的那些成人gbm共享基因特征。多形性黄色星细胞瘤(pxa)是表面、低级星形细胞肿瘤,其主要影响年轻成人。虽然这些肿瘤具有奇异的组织学外观,它们一般是可以适应手术治愈的缓慢生长的肿瘤。然而,某些pxa可以作为gbm复发。纤维状细胞型星形细胞瘤是最常见的儿童期星形细胞肿瘤并且在临床上和组织病理学上区别于影响成人的弥漫性、纤维性星形细胞瘤。纤维状细胞型星形细胞瘤不具有与弥漫性、纤维性星形细胞瘤相同的基因组变化。室管膜下巨细胞星形细胞瘤(sega)是室周、低级星形细胞肿瘤,其通常与结节性硬化(ts)有关并且在组织学上相当于顺ts患者室排列的所谓"蜡烛沟(candle-gutterings)"。类似ts中的其它肿瘤性损伤,它们是缓慢生长的并且可以更近似错构瘤而非真性瘤。婴儿期的促结缔组织增生型大脑星形细胞瘤(dcai)和促结缔组织增生型婴儿神经节神经胶质瘤(digg)是大、表面、通常囊性的良性星形细胞瘤,其在生命的第一年或第二年影响儿童。
1270、少突神经胶质瘤和少突星形细胞瘤(混合性神经胶质瘤)是弥漫性、通常大脑的肿瘤,其在临床上和生物学上最密切相关于弥漫性、纤维性星形细胞瘤。然而,该肿瘤远不及星形细胞瘤常见并且一般具有比弥漫性星形细胞瘤更佳的预后。少突神经胶质瘤和少突星形细胞瘤可以发展为who iii级间变性少突神经胶质瘤或间变性少突星形细胞瘤,或者发展为who iv级gbm。从而,导致少突神经胶质肿瘤的基因变化构成达到gbm的又一途径。
1271、室管膜瘤是临床上多样类型的胶质瘤,其从儿童的侵袭性脑室内肿瘤变化至成人的良性脊髓肿瘤。室管膜瘤向gbm的转变是罕见的。脉络丛肿瘤也是变化类型的优先发生于脑室系统的肿瘤,其从儿童的侵袭性幕上脑室内肿瘤变化至成人的良性小脑脑桥角肿瘤。脉络丛肿瘤有时在li-fraumeni综合征和von hippel-lindau(vhl)疾病患者中有报告。
1272、成神经管细胞瘤是发生在后颅窝的高度恶性、原始肿瘤,其主要发生在儿童中。成神经管细胞瘤是最常见儿童期恶性脑肿瘤。最致死成神经管细胞瘤亚型展示gabaa受体α5亚单位基因的高表达和myc放大。参见例如,j biomed nanotechnol.2016jun;12(6):1297-302。
1273、脑膜瘤是常见的颅内肿瘤,其发生在脑膜中并挤压下方的脑。脑膜瘤通常是良性的,但是某些"非典型"脑膜瘤可以局部复发,并且某些脑膜瘤是明显恶性的并且可以侵入脑或转移。非典型和恶性的脑膜瘤并不如良性脑膜瘤常见。神经鞘瘤是发生在外周神经的良性肿瘤。神经鞘瘤可以发生在颅神经、特别是第八颅神经的前庭部分(前庭神经鞘瘤,听神经瘤),它们在其中呈现为小脑脑桥角团块。成血管细胞瘤是不确定来源的肿瘤,其由内皮细胞,周细胞和所谓的基质细胞构成。这些良性肿瘤最频繁地发生于年轻成人的小脑和脊髓中。多成血管细胞瘤是von hippel-lindau病(vhl)的特征。血管外皮细胞瘤(hpcs)是硬脑膜肿瘤,其可以显示局部侵袭行为并且可以转移。基于硬脑膜的血管外皮细胞瘤(hpc)的组织发生有长久的争论,某些作者将其分类为单独类型而其他将其分类为脑膜瘤的亚型。
1274、本发明提供用于治疗脑癌(例如如本文描述的脑肿瘤)的方法和组合物,其使用含有α5的gabaa受体正变构调节剂,比如选自如本文描述的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在某些实施方式中,治疗包括预防或减缓脑癌进展。在某些实施方式中,治疗包括减轻,改善或减缓与脑癌有关的一种或多种症状的进展。在某些实施方式中,待治疗的症状是认知损害。例如,本公开的方法和组合物能够用来治疗认知损害和/或改善脑癌患者的认知功能。在本发明的某些实施方式中,提供保持或改善脑癌受试者认知功能的方法,所述方法包括下述步骤:给所述受试者施用治疗有效量的本发明化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合。在某些实施方式中,脑肿瘤是成神经管细胞瘤。
1275、研究领域标准(rdoc)
1276、本发明还提供了使用如本文所述的含有α5的gabaa r正变构调节剂或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合治疗神经病学病症和神经精神病病症中的损害的方法和组合物。在某些实施方案中,治疗包括减轻,改善或减慢与所述损害相关的一种或多种症状的发展。在本发明的另一个方面,提供了使用本发明的化合物或其药学上可接受的盐,水合物,溶剂化物,多晶型物,异构体或组合在有此需要的受试者中保留或改善认知功能的方法和组合物。
1277、预见到研究领域标准(rdoc)会增进用于诊断影响神经系统的疾病和障碍的临床标准,诸如dsm和icd(参见,例如,am.j.psychiatry167:7(2010))。rdoc意欲提供基于在基因组学和神经科学以及临床观察中的发现的分类。含有α5的gabaa受体在神经系统中的特定神经回路中的高表达可以是在rdoc下鉴定的神经回路功能障碍的治疗靶标。
1278、关于gabaaα5亚基结合和受体正变构调节剂活性的测定
1279、使用本领域已知的受体结合测定,可以确定受试化合物对包含gabaaα5亚基的gabaa受体的亲和力。参见,例如,美国专利7,642,267和美国专利6,743,789,它们通过引用并入本文。
1280、通过本领域已知的电生理学方法,可以测试受试化合物作为含有α5的gabaa r正变构调节剂的活性。参见,例如,美国专利7,642,267和guidotti等人,psychopharmacology180:191-205,2005。例如,通过测定gaba诱导的包含gabaaα5亚基的gabaa受体的氯离子电导,可以测试正变构调节剂活性。可以使表达这种受体的细胞暴露于有效量的本发明的化合物。可以如下使这种细胞在体内接触本发明的化合物:通过接触含有所述化合物的体液,例如通过接触脑脊髓液。通过使细胞在gaba存在下接触本发明的化合物,可以进行体外试验。在受试化合物存在下表达包含gabaaα5亚基的gabaa受体的细胞中增加的gaba诱导的氯离子电导可以指示所述化合物的正变构调节剂活性。例如,使用对注射了gabaa受体亚基mrna(包括gabaaα5亚基rna)的爪蟾(xenopus)卵母细胞,用编码gabaa受体亚基的质粒转染的hek 293细胞或体内,离体或培养的神经元进行的电压钳测定,可以检测这种电导改变。
1281、本领域普通技术人员应该理解,可以修改和修饰本文所述的方法以适于所针对的应用,并且本文所述的方法可用于其它适合的应用,而且这样的其它添加和改变不会偏离其范围。
1282、从随后的实施例将会更好地理解本发明。但是,本领域技术人员将容易地认识到,讨论的具体方法和结果仅仅是在下文实施方案中更完整地描述的发明的示例。
1283、实施例1:化合物1的合成
1284、
1285、方案11.
1286、
1287、在0℃向5-甲氧基-2-硝基苯胺(5g,29.7mmol)在hcl(浓39ml)中的搅拌混合物中逐滴加入nano2(2.05g,29.7mmol)在h2o(19ml)中的溶液。使内部温度保持低于10℃。加入以后,将该混合物在室温搅拌1小时。将重氮盐通过过滤收集,并用在下一步中。在室温在快速搅拌下向在结晶盘中的重氮盐中逐滴加入nan3(1.93g,29.6mmol)在h2o(7ml)中的溶液。气体产生停止(3小时)后,将其过滤。将收集的固体从meoh中重结晶以得到4.342g(收率75%,2步)作为黄色固体的产物13。在室温向叠氮苯13(1.94g,10mmol)和1,3-丙酮-二甲酸二乙酯(2.20ml,12mmol)在etoh(40ml)中的混合物中加入et3n(1.67ml,12mmol)。将该混合物在室温搅拌60h后,初始的悬浮液变成澄清的黄色溶液。将溶液在真空中浓缩,并将残余物通过色谱法(redisep 24g硅胶柱,10%至40%的etoac在己烷类中的溶液)纯化以得到2.905g作为黄色固体的三唑14。ms:[m+1]=379。
1288、将含有pd/c(10重量%,407mg,0.38mmol)的以上三唑14(2.95g,7.66mmol)在etoh(50ml)中的溶液在h2(气球)下搅拌24小时。将其穿过硅藻土过滤。将滤液浓缩,并将残余物通过色谱法(redisep 24g硅胶柱,10%至50%的etoac在己烷类中的溶液)纯化以得到2.453g作为白色固体的苯胺15。(70%收率,2步)。ms:[m+1]=349。
1289、将化合物15(2.45g,7.03mmol)和催化量的p-tsoh·h2o(24mg)在对二甲苯(30ml)中在140℃油浴中加热过夜。将该混合物冷却并过滤。将该固体用冷的etoac洗涤。干燥后,得到1.88g(88%收率)的内酰胺16。ms:[m+1]=303。
1290、在室温向内酰胺酯16(837mg,2.77mmol)在thf(20ml)中的悬浮液中加入libh4(2m的在thf中的溶液,1.39ml,2.78mmol)。将该混合物在室温搅拌60h后,加入更多的libh4(2m的在thf中的溶液,0.28ml,0.56mmol),并将其在室温再搅拌24小时。将etoac/etoh(10ml/10ml)的混合物加入该反应中,并将其在真空中浓缩。将残余物溶解于etoac/ch2cl2/meoh中,并加入疏松的硅胶。将挥发性溶剂蒸发后,将固体加载到redisep 24g硅胶柱上。色谱法(溶剂a:etoac,溶剂b:10:1v/v ch2cl2/meoh;梯度洗脱液:a至b)得到540mg(75%收率)作为白色固体的醇17。ms:[m+1]=261。
1291、历时20分钟向醇17(105.4mg,0.40mmol)和cbr4(336mg,1.01mmol)在dmf(3ml)中的溶液中缓慢地加入pph3(255mg,0.97mmol)在dmf(1ml)中的溶液。加入以后,tlc指示该反应完成。将水加入以猝灭该反应,并将该混合物用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 12g硅胶柱,ch2cl2至30%的etoac在ch2cl2中的溶液)得到439.2mg溴化物18([m+1]=324)和ph3po的混合物。将含有pd/c(10重量%,200mg,0.19mmol)的在etoac/etoh(8ml/8ml)中的以上混合物(439mg)在h2(气球)下搅拌2小时,然后穿过硅藻土过滤。将滤液浓缩,并将残余物通过色谱法(redisep 12g硅胶柱,溶剂a:1:1v/vch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:a至b)纯化以得到99mg(~80%收率,2步)作为白色固体的产物19。ms:[m+1]=245。
1292、在一个单独的烧瓶中,在0℃将在ch3cn(1ml)中的1,2,3-三唑(55.3mg,0.80mmol)用i-pr2net(146μl,0.84mmol)处理,随后用pocl3(23μl,0.25mmol)处理。将该溶液在0℃搅拌2小时。在一批次中加入内酰胺19,并将得到的悬浮液在80℃油浴中加热20小时。将水加入以猝灭该反应。将其用etoac萃取三次。将合并的萃取物用盐水洗涤,并经na2so4干燥。过滤并浓缩,得到48.8mg粗产物20,将其直接用在下一步中。将ko-t-bu(37.2mg,0.33mmol)在dmf(0.5ml)中的溶液冷却至-50℃。逐滴加入异氰基乙酸乙酯(40μl,0.36mmol)。将该混合物在-50℃搅拌1小时。逐滴加入在dmf(1ml)中的以上粗产物20。使该混合物温热至10℃,并在10℃搅拌1小时。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物依次用水,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物。
1293、色谱法(redisep 12g硅胶柱,溶剂a:1:1v/v ch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:20%至80%的b在a中的溶液)得到15mg(21%收率,2步)作为灰白色固体的化合物1(实施例1)。ms:[m+1]=340。1h-nmr(500mhz,cdcl3)δ:7.74(s,1h),7.63(d,1h,j=3hz),7.51(d,1h,j=8.5hz),7.14(dd,1h,j=3.0,8.5hz),4.44(q,2h,j=7.0hz),3.95(s,3h),2.44(s,3h),1.45(t,3h,j=7.0hz)。
1294、实施例2:化合物2的合成:
1295、
1296、以与关于实施例1描述的途径类似的合成途径,使用5-氟-2-硝基-苯胺作为起始原料,合成实施例2的化合物,以得到作为浅棕色固体的化合物2:ms:[m+1]=328。1h-nmr(500mhz,cdcl3)δ:7.90(br dd,1h,j=2.5,8.5hz),7.77(s,1h),7.62(br dd,1h,j=5.0,9.0hz),7.35(m,1h),4.45(q,2h,j=7.0hz),2.45(s,3h),1.45(t,3h,j=7.0hz)。
1297、实施例3:化合物3的合成:
1298、
1299、以与关于实施例1描述的途径类似的合成途径,使用2-硝基-苯胺作为起始原料,合成实施例3的化合物,以得到作为浅黄色固体的化合物3:ms:[m+1]=310;1h-nmr(500mhz,cdcl3)δ:8.161(br d,1h,j=8.5hz),7.81(s,1h),7.66(m,3h),4.45(q,2h,j=7.0hz),2.45(s,3h),1.46(t,3h,j=7.0hz)。
1300、实施例4:化合物110的合成
1301、
1302、在使用前将乙酰胺肟在甲苯中共沸三次。向乙酰胺肟(30mg,0.4mmol)在thf(1ml)中的悬浮液中加入60%的nah在油中的分散体(16mg,0.4mmol)。将该悬浮液在室温搅拌15分钟。加入酯化合物2(65mg,0.2mmol)。将含该酯的小瓶用加入该反应混合物中的thf(1ml)漂洗。将得到的棕色悬浮液在室温搅拌30分钟,然后在70℃加热2小时30分钟。将该悬浮液用meoh猝灭。将溶剂蒸发,并将粗制油状物通过色谱法(redisep 4g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)纯化以得到28mg(41%收率)产物。ms:[m+1]=338。h1nmr(cdcl3)δ7.92(1h,dd,j=2.5,8.5hz),7.90(1h,s),7.67(1h,dd,j=4.5,9.5hz),7.38(1h,m),2.51(3h,s),2.46(3h,s)。
1303、实施例5:化合物167的合成
1304、
1305、所述化合物类似地从化合物1制备以得到化合物167:ms:[m+1]=350。h1nmr(cdcl3)δ7.87(1h,s),7.65(1h,d,j=3hz),7.55(1h,d,j=9hz),7.17(1h,dd,j=2.5,9hz),3.96(3h,s),2.5(3h,s),2.45(3h,s)。
1306、方案12.
1307、
1308、实施例6:化合物4的合成:
1309、
1310、向如实施例1中制备的化合物17(260mg)在dmso(4ml)和ch2cl2(6ml)中的溶液中加入et3n(0.7ml,5mmol),随后加入py·so3(398mg,2.5mmol)。将其在室温搅拌1小时。将该反应混合物倒入水中,并用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到198.5mg粗制醛21,将其不经进一步纯化地使用。在0℃向醛21(198.5mg,0.77mmol)在thf(10ml)中的悬浮液中逐滴加入phmgbr(1m的在thf中的溶液,1.54ml,1.54mmol)。将其在0℃搅拌30分钟。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。
1311、将合并的萃取物用盐水洗涤,并经na2so4干燥。过滤并浓缩,得到252.9mg作为棕色泡沫状固体的苯甲醇22。将其不经进一步纯化地用于下一步。向含有et3sih(0.60ml,3.76mmol)的以上粗制醇22在ch2cl2(8ml)中的溶液中加入tfa(0.64ml,8.27mmol)。将该反应溶液在室温搅拌4小时。浓缩后,将残余物通过色谱法(redisep 12g硅胶柱,20%至80%的etoac在己烷类中的溶液)纯化以得到34.1mg(收率12%,4步)作为白色泡沫状固体的还原的产物23。ms:[m+1]=321。
1312、在一个单独的烧瓶中,在0℃将1,2,4-三唑(27mg,0.39mmol)在ch3cn(0.5ml)中的溶液用i-pr2net(72μl,0.41mmol),随后用pocl3(11μl,0.12mmol)处理。将该混合物在0℃搅拌2小时。将内酰胺物质23(32.2mg,0.1mmol,固体)在一批次中加入该反应混合物中,并将其在80℃油浴中加热20小时。将该混合物冷却至室温,观察到奶油状固体沉淀物。加入水(0.5ml),并将其在室温搅拌5分钟。将固体沉淀物通过过滤收集,并用0.5ml的水洗涤,随后在高真空下干燥,以得到15.8mg(收率42%)作为灰白色蓬松固体的加成化合物24。ms:[m+1]=372。将ko-t-bu(9.5mg,85μmol)在dmf(0.5ml)中的溶液冷却至-50℃。逐滴加入异氰基乙酸乙酯(10.4μl,95μmol)。将得到的混合物在-50℃搅拌1小时。在一批次中加入三唑脒24(15.8mg,42μmol,固体)。在1h内将搅拌混合物温热至10℃,并在10℃保持1小时。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱。溶剂a:1:1v/v ch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:a至50%的b在a中的溶液)得到16.8mg(收率95%)作为白色固体的实施例6的化合物。ms:[m+1]=416。1h-nmr(500mhz,cdcl3)δ:7.74(s,1h),7.63(d,1h,j=3.0hz),7.50(d,1h,j=9.0hz),7.30(br d,2h,j=7.0hz),7.29(br d,2h,7.5hz),7.20(m,1h),7.13(dd,1h,j=2.5,9.0hz),4.41(q,2h,j=7.5hz),4.17(s,2h),3.95(s,3h),1.43(t,3h,7.5hz)。
1313、实施例7:化合物5的合成:
1314、
1315、以与关于实施例6描述的途径类似的合成途径,使用2-硝基-苯胺作为起始原料,合成实施例7的化合物,以得到作为棕色固体的化合物5:ms:[m+1]=386。1h-nmr(500mhz,cdcl3)δ:8.16(br d,1h,j=7.0hz),7.81(s,1h),7.60-7.68(m,3h),7.34(br d,2h,j=8.0hz),7.29(br d,2h,j=7.0hz),7.20(m,1h),4.42(q,2h,j=7.0hz),4.18(s,2h),1.44(t,3h,j=7.0hz)。
1316、实施例8:化合物6的合成:
1317、
1318、以与关于实施例6描述的途径类似的合成途径,使用5-氟-2-硝基-苯胺作为起始原料,合成实施例8的化合物,以得到作为棕色固体的化合物8:ms:[m+1]=404。1h-nmr(500mhz,cdcl3)δ:7.90(dd,1h,j=3.5,8.5hz),7.77(s,1h),7.61(dd,1h,j=5.0,10.5hz),7.28-7.37(m,5h),7.21(m,1h),4.43(q,2h,j=7.0hz),4.17(s,2h),1.44(t,3h,j=7.0hz)。
1319、实施例9:化合物44的合成:
1320、
1321、以与关于实施例6描述的途径类似的合成途径,使用5-氟-2-硝基-苯胺作为起始原料,合成实施例9的化合物,以得到作为淡棕色固体的实施例9的化合物:ms:[m+1]=418。1h-nmr(500mhz,cdcl3)δ:7.89(br d,1h,j=9.5hz),7.76(s,1h),7.60(dd,1h,j=5.5,10.0hz),7.35(br t,1h,j=6.0hz),7.22(br d,2h,j=8.5hz),7.09(br d,2h,j=7.5hz),4.43(q,2h,j=7.5hz),4.12(s,2h),2.30(s,3h),1.44(t,3h,j=7.5hz)。
1322、实施例10:化合物45的合成:
1323、
1324、以与关于实施例6描述的途径类似的合成途径,使用5-氟-2-硝基-苯胺作为起始原料,合成实施例10的化合物,以得到作为淡棕色固体的实施例10的化合物:ms:[m+1]=438。1h-nmr(500mhz,cdcl3)δ:7.90(dd,1h,j=3.0,8.0hz),7.77(s,1h),7.61(dd,1h,j=5.0,9.0hz),7.36(m,1h),7.25(br s,4h),4.42(q,2h,j=7.0hz),4.14(s,2h),1.44(t,3h,j=7.0hz)。
1325、实施例11:化合物46的合成:
1326、
1327、以与关于实施例6描述的途径类似的合成途径,使用5-氟-2-硝基-苯胺作为起始原料,合成实施例11的化合物,以得到作为微黄色固体的实施例11的化合物:ms:[m+1]=422。1h-nmr(500mhz,cdcl3)δ:7.90(dd,1h,j=3.0,8.5hz),7.77(s,1h),7.61(dd,1h,j=5.0,9.0hz),7.36(m,1h),7.28(m,2h),6.96(m,2h),4.42(q,2h,j=7.5hz),4.14(s,2h),1.44(t,3h,j=7.0hz)。
1328、实施例12:化合物47的合成:
1329、
1330、以与关于实施例6描述的途径类似的合成途径,使用2-硝基-苯胺作为起始原料,合成实施例12的化合物,以得到作为微黄色固体的实施例12的化合物:ms:[m+1]=420。1h-nmr(500mhz,cdcl3)δ:8.16(br d,1h,j=7.0hz),7.80(s,1h),7.64(m,3h),7.25(m,4h),4.41(q,2h,j=7.0hz),4.14(s,2h),1.44(t,3h,j=8.0hz)。
1331、实施例13:化合物109的合成:
1332、
1333、将乙酰胺肟(50mg,0.67mmol)与甲苯共沸3次。加入thf(5ml),然后加入60%的nah在油中的分散体(25mg,0.62mmol)。将该悬浮液在室温搅拌30分钟。将2ml的该悬浮液加入酯化合物6(40mg,0.099mmol)中,并将得到的溶液在70℃加热3h。将该溶液用水猝灭。将该溶液用etoac(3×)萃取。将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 12g硅胶柱,用50%的etoac在己烷类中的溶液洗脱)得到6mg(收率20%)作为黄色固体的产物化合物109。ms:[m+1]=414)。h1nmr(cdcl3)δ7.93(1h,dd,j=3,8.5hz),7.89(1h,s),7.65(1h,dd,j=5.5,9hz),7.38(1h,m),7.23(5h,m),4.2(2h,s),2.50(3h,s)。
1334、实施例14:化合物7的合成:
1335、
1336、在0℃向5-甲氧基-2-硝基苯胺(5g,29.7mmol)在hcl(浓12.9ml)中的搅拌混合物中逐滴加入nano2(2.05g,29.7mmol)在h2o(8ml)中的溶液。使内部温度保持低于5℃。加入以后,在1h内将该混合物温热至室温。将该混合物冷却至0℃,并缓慢滴加sncl2·2h2o(20.13g,89.2mmol)在hcl(浓13ml)中的溶液。加入以后,将其在室温搅拌2小时。将得到的黄色固体通过过滤收集,并用冷的(0℃)6n hcl洗涤。在真空干燥箱中干燥后,得到3.245g(收率50%)作为棕色固体的芳基肼25。ms:[m+h2o+na]=224。在一个单独的烧瓶中,将1,3-丙酮二甲酸二乙酯(2.426g,12mmol)和乙酸二乙氧基甲酯(1.946g,12mmol)的混合物在微波辐射下在100℃加热1小时。将该反应混合物在真空中浓缩,并将残余的挥发性组分与甲苯(5ml)一起在真空中共蒸馏出,以得到缩合产物26,将其直接用在下一步中。
1337、方案13.
1338、
1339、将来自以上步骤的产物26溶解在etoh(30ml)中。加入分子筛(2g)和盐酸肼25(2.19g,10mmol)。将该悬浮液在室温搅拌24小时。将其穿过硅藻土过滤,并将该固体用etoac(10ml x 3)洗涤。将滤液浓缩。将残余物通过色谱法(redisep 40g硅胶柱,10%至40%的etoac在己烷类中的溶液)纯化以得到2.091g吡咯27,将其不经进一步纯化地用于下一步。ms:[m+1]=378。
1340、将以上在27(2.09g,5.5mmol)上的硝基在etoh(40ml)中用pd/c(10重量%,295mg,0.28mmol)在h2(气球)下还原18小时。将该混合物穿过硅藻土过滤。将滤液浓缩,并将残余物通过色谱法(redisep 24g硅胶柱,己烷类至50%的etoac在己烷类中的溶液)纯化以得到1.127g作为黄色粘性油的未环化产物28([m+1]=348),和154mg作为灰色固体的环化产物29(ms:[m+1]=302)。将在对二甲苯(20ml)中的未环化的苯胺28(1.127g,3.2mmol)用催化量的p-tsoh·h2o(15mg)在140℃油浴中处理20小时。将该反应混合物冷却,浓缩,并将残余物与冷的(0℃)etoac一起研磨。过滤得到559mg作为黄色固体的内酰胺产物29。合并的内酰胺产物29的总重量是713mg(24%,3个步骤)。ms:[m+1]=302。
1341、在-78℃向酯29(566mg,1.88mmol)在ch2cl2(35ml)中的悬浮液中加入dibal-h(1m在己烷中,6.60ml,6.60mmol)。将该悬浮液在-78℃搅拌10分钟。移去冷却浴,并将其搅拌20分钟,同时温度升至室温。这时,tlc指示~80%反应完成。将其冷却至-78℃,并加入更多的dibal-h(1m在己烷中,1.0ml,1.0mmol)。在-78℃搅拌30分钟后,lcms指示反应进行至完成。通过加入罗谢尔盐水溶液(20%),随后加入etoac将该反应猝灭。将其在室温剧烈搅拌直至其变为澄清的两层混合物。将各层分离,并将水层用etoac萃取三次。将合并的有机相用盐水洗涤并经na2so4干燥。过滤并浓缩,得到480mg作为淡黄色固体的粗制醇30。ms:[m+1]=260。
1342、在30分钟内向醇30(200mg,0.77mmol)和cbr4(640mg,1.93mmol)在dmf(8ml)中的溶液中缓慢地加入pph3(486mg,1.85mmol)在dmf(2ml)中的溶液。加入以后,将其在室温搅拌30分钟。将水加入以猝灭该反应,并将该混合物用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物。色谱法(redisep12g硅胶柱,溶剂a:1:1v/v ch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:10%至40%的b在a中的溶液)得到221mg溴化物31和ph3po的混合物。
1343、将含有pd/c(10重量%,200mg,0.19mmol)的在etoac/etoh(8ml/8ml)中的以上混合物在h2(气球)下搅拌1小时。将其穿过硅藻土过滤。将滤液浓缩,并将残余物通过色谱法(redisep 12g硅胶柱,溶剂a:1:1v/v ch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:10%至40%的b在a中的溶液)纯化以得到146mg还原产物32([m+1]=244)和ph3po的混合物。
1344、在一个单独的烧瓶中,将1,2,4-三唑(81mg,1.17mmol)在ch3cn(1ml)中的溶液在0℃用i-pr2net(214μl,1.23mmol),随后用pocl3(34μl,0.36mmol)处理。将该溶液在0℃搅拌2小时。在一批次中加入内酰胺32(lcms指示~60%纯度),并将得到的悬浮液在80℃油浴中加热18小时。将水加入以猝灭该反应。将其用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到126.6mg作为黄色胶状物的粗产物33,将其直接用于下一个反应中。ms:[m+1]=295。将ko-t-bu(97mg,0.86mmol)在dmf(1ml)中的溶液冷却至-50℃。逐滴加入异氰基乙酸乙酯(104μl,0.95mmol)。将该混合物在-50℃搅拌1小时。逐滴加入在dmf(1.5ml)中的以上粗产物33。使该混合物温热至10℃,并在10℃搅拌1小时。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物依次用水,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 12g硅胶柱,溶剂a:1:1v/vch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:10%至40%的b在a中的溶液)得到22mg白色固体,将其通过制备型tlc(用1:1a/b显影)进一步纯化,得到12.8mg作为白色固体的终产物化合物7(实施例14)。ms:[m+1]=339。1h-nmr(500mhz,cdcl3)δ:7.70(s,1h),7.56(s,1h),7.50(d,1h,j=3.0hz),7.43(d,1h,j=8.5hz),7.00(dd,1h,j=2.5,9.5hz),5.29(br s,1h),4.44(q,2h,j=7.0hz),3.92(s,3h),3.55(br s,1h),2.17(s,3h),1.45(t,3h,j=7.0hz)。
1345、实施例15:化合物8的合成:
1346、
1347、方案14.
1348、
1349、向在实施例14中制备的醇30(261mg,1.0mmol)在dmso(4ml)和ch2cl2(6ml)中的溶液中加入et3n(0.7ml,5mmol),随后加入py·so3(398mg,2.5mmol)。将其在室温搅拌1小时。将该反应混合物倒入水中,并用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤并浓缩,得到226mg作为黄色固体的粗制醛34。将其未经纯化地用于下一步。ms:[m+1]=258。
1350、在0℃向粗制醛34(202mg,0.79mmol)在thf(10ml)中的悬浮液中逐滴加入phmgbr(1m的在thf中的溶液,1.58ml,1.58mmol)。将其在0℃搅拌30分钟。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物用盐水洗涤,并经na2so4干燥。过滤并浓缩,得到275mg作为黄色泡沫状固体的粗产物35,将其未经纯化地用于下一步中。
1351、向含有et3sih(0.66ml,4.10mmol)的以上的粗制醇35在ch2cl2(10ml)中的溶液中加入tfa(0.70ml,9.02mmol)。将该反应溶液在室温搅拌1小时。浓缩后,将残余物通过色谱法(redisep 24g硅胶柱,10%至50%的etoac在己烷类中的溶液)纯化以得到187.8mg(收率59%,3个步骤)作为灰色固体的产物36。ms:[m+1]=320。
1352、在一个单独的烧瓶中,在0℃将1,2,4-三唑(127mg,1.83mmol)在ch3cn(1.6ml)中的溶液用i-pr2net(336μl,1.93mmol),随后用pocl3(53μl,0.56mmol)处理。将该混合物在0℃搅拌2小时。将内酰胺36(150mg,0.47mmol,固体)在一批次中加入该反应混合物中,并将其在80℃油浴中加热18小时。将该混合物冷却至室温,并观察到固体沉淀物。加入水(2.1ml),并将其在室温搅拌10分钟。过滤,将固体用2ml的水洗涤,随后在高真空下干燥,得到118.8mg(收率69%)作为灰白色蓬松固体的三唑脒37。ms:[m+1]=371。将ko-t-bu(72mg,0.64mmol)在dmf(2ml)中的溶液冷却至-50℃。逐滴加入异氰基乙酸乙酯(77μl,0.71mol)。将得到的混合物在-50℃搅拌1小时。将三唑脒37(118.8mg,42μmol,固体)一批加入。在1h内将搅拌混合物温热至10℃,并在10℃保持1小时。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物依次用h2o,盐水洗涤,并经na2so4干燥。过滤,浓缩,然后色谱法(redisep 12g硅胶柱。溶剂a:1:1v/v ch2cl2/己烷类,溶剂b:etoac;梯度洗脱液:a至40%的b在a中的溶液)得到125.1mg(收率94%)作为白色固体的化合物8。ms:[m+1]=415。1h-nmr(500mhz;cdcl3)δ:7.72(s,1h),7.54(s,1h),7.51(br s,1h),7.44(br d,1h,j=9.5hz),7.29(br d,2h,j=7.5hz),7.20(m,3h),7,01(br d,1h,j=7.5hz),5.30(br s,1h),4.38(q,2h,j=7.0hz),3.92(br s,5h),3.54(br s,1h),1.41(t,3h,j=7.0hz)。
1353、实施例16:化合物9的合成:
1354、
1355、方案15.
1356、
1357、在室温将lioh(1.09g,45.5mmol)加入酯16(在实施例1中制备)(2.75g,9.10mmol)在thf(24ml)和水(20ml)中的搅拌溶液中。加入meoh(4ml),并在室温继续搅拌2小时,这时lcms指示酯完全消耗。在真空中浓缩后,通过加入2n hcl(20ml)将该反应混合物酸化至ph3-4。搅拌20分钟后,将该反应混合物冷却至0℃,将固体沉淀物通过过滤收集,用3-4ml水洗涤,并干燥,得到1.59g(64%)作为浅灰色固体的对应酸38。ms:[m+1]=275。向在dcm(30ml)中混悬并搅拌的酸38(1.59g,5.8mmol)中加入edc(5.6g,29.2mmol),苯甲醇(2.5g,23.2mmol)和dmap(3.54g,29.2mmol)。在室温搅拌3天后,将该反应混合物在真空中浓缩。将水(80ml)加入该浆料中,随后加入乙醚(40ml),并将该混合物剧烈搅拌40分钟,这时该浆料变成沉淀,通过吸滤收集。将该固体用水和少量的乙醚洗涤,并干燥,得到1.65g(78%)作为白色固体的苄酯39。ms:[m+1]=365。
1358、在0℃将化合物1,2,4-三唑(1.22g,17.7mmol)在ch3cn(15ml)中的溶液用i-pr2net(3.24ml,18.6mmol),随后用pocl3(0.507ml,5.44mmol)处理。将该溶液在0℃搅拌2小时。将苄酯39(1.65g,4.53mmol)一批加入,并将得到的悬浮液在80℃油浴中加热18小时。lcms指示剩余5-10%的起始内酰胺。在一个单独的烧瓶中,将在ch3cn(3.8ml)中的1,2,4-三唑(307mg,总共4.9当量)用i-pr2net(0.82ml,总共5.1当量)和pocl3(0.127ml;总共1.5当量)在0℃处理2小时。将得到的澄清溶液转移至以上反应混合物。在80℃加热2h后,将该反应冷却至室温,将水缓慢地加入以猝灭该反应(10分钟)。在冰浴中冷却后,将形成的固体通过过滤收集,用水(5ml)洗涤,并干燥,以得到1.61g(86%)作为浅黄色固体的产物40。ms:[m+1]=416。
1359、将ko-t-bu(0.739g,6.59mmol)在dmf(11ml)中的溶液冷却至-50℃。逐滴加入异氰基乙酸乙酯(0.810ml,7.00mmol)。将该混合物在-50℃搅拌1小时。加入以上三唑中间体40(1.61g,3.87mmol)。将该混合物在-50℃搅拌30分钟,并历时4-5h缓慢地温热至室温。加入饱和的nh4cl水溶液(10ml),随后加入etoac(10ml)。将该混合物声处理以分解固体块,然后彻底搅拌30分钟。将沉淀物通过过滤收集,用水,et2o洗涤,并干燥,以得到作为白色固体的粗产物。将滤液在水和etoac之间分配;将水层分离,并用etoac萃取两次;将合并的etoac层用盐水洗涤并经mgso4干燥。过滤,并除去溶剂,得到固体残余物,将其与上文得到的固体合并用于色谱纯化(使用redisep 24g硅胶柱,用0.5-5%的meoh在dcm中的溶液梯度洗脱),得到1.78g(100%)作为白色固体的咪唑41。ms:[m+1]=460。将苄酯41(1.78g,3.87mmol)在催化量的10%炭载pd存在下,在thf(40ml),meoh(20ml)和etoac(20ml)的溶剂混合物中氢解(氢气球)20小时。lcms指示起始原料完全消失。将固体催化剂通过硅藻土过滤除去,并用足量的30%的meoh在dcm中的溶液重复漂洗直至回收几乎所有产物(tlc监测)。将含产物的滤液在真空中浓缩,得到1.22g(85%)作为微黄色固体的酸产物42。ms:[m+1]=370。
1360、在0℃向在thf(25ml)中混悬并搅拌的酸42(1.22g,3.30mmol)中滴加硼烷二甲硫醚复合物(2m thf;19ml,38mmol)。除去冰浴,并将该反应混合物在室温搅拌16小时。在冰浴中冷却后,将该反应用meoh(20ml)小心地猝灭,然后在室温搅拌过夜。在真空中除去溶剂。再进行两次meoh的加入和在真空中除去。使用1-8%的meoh在dcm中的溶液梯度的isco纯化(redisep 24g柱)得到0.625g(53%)作为白色固体的醇产物43。ms:[m+1]=356。
1361、在0℃将偶氮二甲酸二异丙酯(48.3mg,0.233mmol)逐滴加入醇43(37.5mg,0.106mmol),苯酚(14.9mg,0.158mmol)和ph3p(55.6mg,0.212mmol)在无水thf(0.8ml)中的搅拌溶液中。除去冰浴,并在室温继续搅拌16小时。lcms指示起始醇完全消失。将该反应混合物在饱和nahco3和etoac之间分配。将有机层分离,并用水,盐水洗涤,并经mgso4干燥。通过两个连续制备型tlc(4%的meoh在dcm中的溶液和己烷类/etoac/meoh=47.5/47.5/5,v/v/v)从该反应混合物分离期望产物,得到5.3mg(12%)作为白色固体的产物,其为化合物9。ms:[m+1]=432。1h-nmr(500mhz,cdcl3)δ:7.77(s,1h),7.63(d,1h,j=3.5hz),7.53(d,1h,j=9.0hz),7.31(m,2h),7.17(dd,1h,j=3.0,8.5hz),7.08(d,2h,j=7.0hz),6.99(t,1h,j=6.5hz),5.30(s,2h),4.40(q,2h,j=7.0hz),3.96(s,3h),1.38(t,3h,j=7.0hz)。
1362、实施例17:化合物10的合成:
1363、
1364、以与关于实施例16描述的途径类似的合成途径,在最终的步骤中使用4-氟-苯酚,合成实施例17的化合物,以得到作为白色固体的化合物10(4.9mg):ms:[m+1]=450。1h-nmr(500mhz,cdcl3)δ:7.76(s,1h),7.64(d,1h,j=3.5hz),7.53(d,1h,j=8.0hz),7.17(dd,1h,j=2.5,8.0hz),7.01(m,4h),5.26(s,2h),4.40(q,2h,j=7.0hz),3.96(s,3h),1.40(t,3h,j=7.0hz)。
1365、实施例18:化合物11的合成:
1366、
1367、以与关于实施例16描述的途径类似的合成途径,在最终的步骤中使用3-甲氧基-苯酚,合成实施例18的化合物,以得到作为白色固体的化合物11(6.1mg):ms:[m+1]=462。1h-nmr(500mhz,cdcl3)δ:7.76(s,1h),7.63(d,1h,j=2.5hz),7.53(d,1h,j=9.0hz),7.15-7.22(m,2h),6.67(m,2h),6.55(br dd,1h,j=2.5,8.0hz),5.28(s,2h),4.39(q,2h,j=7.0hz),3.96(s,3h),3.81(s,3h),1.39(t,3h,j=7.0hz)。
1368、实施例19:化合物12的合成:
1369、
1370、以与关于实施例16描述的途径类似的合成途径,在最终的步骤中使用2,4-二甲基苯酚,合成实施例19的化合物,以得到作为白色固体的化合物12(3.1mg):ms:[m+1]=460。1h-nmr(500mhz,cdcl3)δ:7.76(s,1h),7.65(d,1h,j=3.0hz),7.53(d,1h,j=9.0hz),7.17(dd,1h,j=2.5,8.5hz),6.98(m,3h),5.26(s,2h),4.37(q,2h,j=7.0hz),3.96(s,3h),2.26(s,3h),2.20(s,3h),1.36(t,3h,j=7.0hz)。
1371、实施例20:化合物107的合成:
1372、
1373、向醇43(其中x=f)(以与其中x=och3的实施例相同的方式制备)(60mg,0.17mmol)在thf(0.8ml)中的溶液中加入苯酚(30mg,0.32mmol),三苯基膦(84mg,0.32mmol)。将该反应混合物在室温搅拌15分钟。然后将其用冰浴冷却,并缓慢地加入diad(64μl,0.32mmol)在thf(0.2ml)中的溶液。除去冰浴,并将该反应混合物在室温搅拌18h。lcms指示仍然存在一些起始原料。将苯酚(10mg),三苯基膦(28mg)和diad(21μl)加入该反应混合物中,并搅拌另外1小时。将溶剂蒸发,并将粗制物质通过色谱法(redisep 12g硅胶柱。洗脱溶剂:etoac)和制备型tlc(洗脱溶剂:5%meoh/47.5%etoac/47.5%己烷类)纯化以得到11.4mg(收率16%)产物化合物107。[m+1]=421)。h1nmr(cdcl3)δ7.92(1h,dd,j=3.5,8.5hz),7.80(1h,s),7.63(1h,dd,j=5,10hz),7.38(1h,m),7.31(2h,t,j=8.5hz),7.07(2h,d,j=8.5hz),7.00(1h,t,j=8.5hz),5.3(2h,s),4.39(2h,q,j=7hz),1.38(3h,t,j=7hz)。
1374、实施例21:化合物111的合成:
1375、
1376、向醇43(x=me)(160mg,0.47mmol)在乙腈(9ml)中的悬浮液中加入pobr3(405mg,1.41mmol)。将该反应混合物在80c加热5小时。将该反应混合物用冰浴冷却,并加入饱和的nahco3水溶液。将得到的溶液用dcm(3x)萃取。将合并的有机相用盐水洗涤,并经mgso4干燥。将溶剂浓缩以得到期望产物(166mg,88%收率,[m+1]=403)。
1377、向以上烷基溴衍生物(30mg;0.075mmol)在除氧的dme(2.7ml)中的悬浮液中加入3-吡啶硼酸(14mg,0.11mmol)和2m na2co3溶液(0.22ml,0.44mmol)。将该悬浮液在室温搅拌5分钟,然后加入pdcl2(pph3)2(10mg,0.015mmol)。将该悬浮液在微波中在85c加热1小时。将该反应混合物冷却,用水稀释,并用etoac(两次)萃取。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物,将其通过2个制备型tlc(洗脱系统:3%的meoh在dcm中的溶液)纯化以得到5.3mg(收率18%)产物化合物111。ms:[m+1]=401。h1nmr(cdcl3)δ8.66(1h,bs),8.48(1h,bs),7.96(1h,s),7.79(1h,s),7.66(1h,d,j=8hz),7.50(1h,d,j=8hz),7.43(1h,d,j=7hz),7.23(1h,m),4.42(2h,q,j=7hz),4.18(2h,s),2.54(3h,s),1.44(3h,t,j=7hz)。
1378、实施例22:化合物48的合成:
1379、
1380、方案16.
1381、
1382、在室温向在dmso(1ml)和二氯甲烷(2.5ml)中搅拌的醇43(186mg,0.523mmol)中加入三乙胺(0.394ml,2.82mmol)和吡啶三氧化硫复合物(225mg,1.41mmol)。搅拌3h后,将该反应用水猝灭(5ml),并用乙酸乙酯萃取三次。将合并的有机溶液用水,盐水洗涤,并经mgso4干燥。将醛产物57通过使用0.5-8%的meoh在dcm中的溶液梯度洗脱的isco快速柱色谱法(redisep 4g柱)分离。得到84.4mg(46%)微黄色泡沫状固体。ms:[m+1]=354。
1383、在室温向醛57(15.5mg,0.0439mmol)在1,2-二氯乙烷(0.3ml)中的搅拌溶液中加入吡咯烷(5.5ul,0.0658mmol)。搅拌2分钟后,溶液变为澄清,加入nabh(oac)3(14.4mg)。将该反应混合物搅拌4小时,用饱和的nahco3猝灭,并用乙酸乙酯萃取三次。将合并的有机层用水,盐水洗涤,并经na2so4干燥。使用10%的meoh在dcm中的溶液的制备型tlc产生13.1mg(73%)作为澄清膜样固体的期望化合物48。ms:[m+1]=409。1h-nmr(500mhz,cdcl3)δ:7.74(s,1h),7.62(d,1h,j=3.0hz),7.51(d,1h,j=9.0hz),7.14(dd,1h,j=3.5,9.0hz),4.42(q,2h,j=6.5hz),3.94(s,3h),3.87(br s,2h),2.65(br s,4h),1.79(br s,4h),1.44(t,3h,j=7.0hz)。
1384、实施例23:化合物49的合成:
1385、
1386、以与关于实施例22描述的途径类似的合成途径,在最终的步骤中使用吗啉,合成实施例23的化合物,以得到作为澄清膜样固体的实施例23的化合物:ms:[m+1]=425。1h-nmr(500mhz,cdcl3)δ:7.75(s,1h),7.63(d,1h,j=3.0hz),7.52(d,1h,j=9.5hz),7.15(dd,1h,j=3.0,9.0hz),4.42(q,2h,j=7.5hz),3.95(s,3h),3.76(br s,2h),3.71(br s,4h),2.57(br s,4h),1.44(t,3h,j=8.0hz)。
1387、实施例24:化合物50的合成:
1388、
1389、以与关于实施例22描述的途径类似的合成途径,在最终的步骤中使用二乙胺,合成实施例24的化合物,以得到作为澄清膜样固体的实施例24的化合物:ms:[m+1]=411。1h-nmr(500mhz,cdcl3)δ:7.74(s,1h),7.64(br d,1h,j=3.0hz),7.51(d,1h,j=9.0hz),7.15(dd,1h,j=2.5,9.0hz),4.43(q,2h,j=6.5hz),3.96(s,3h),3.86(br s,2h),2.64(br s,4h),1.44(t,3h,j=8.5hz),1.15(br s,6h)。
1390、实施例25:化合物51的合成:
1391、
1392、以与关于实施例22描述的途径类似的合成途径,在最终的步骤中使用甲基苄基胺,合成实施例25的化合物,以得到作为澄清膜样固体的实施例25的化合物:ms:[m+1]=459。1h-nmr(500mhz,cdcl3)δ:7.75(s,1h),7.63(d,1h,j=3.0hz),7.51(d,1h,j=8.5hz),7.36(br d,2h,j=8.0hz),7.30(m,2h),7.23(m,1h),7.15(dd,1h,j=3.0,9.0hz),4.38(q,2h,j=7.5hz),3.95(s,3h),3.85(br s,2h),3.63(br s,2h),2.25(s,3h),1.41(t,3h,j=7.0hz)。
1393、实施例26:化合物170的合成:
1394、
1395、在圆底烧瓶中将异丁脒肟(isobutyramidoxime)(41.8mg,0.41mmol)和酯48(27.9mg,0.0683mmol)在甲苯中在rotavap上共沸若干次,悬浮于无水thf(0.6ml)中,然后冷却至0℃。加入nah(60%油悬浮液;10.9mg,0.273mmol)。除去冰浴,将该反应混合物在室温搅拌20分钟,随后在70℃加热6小时,并冷却。加入水(4ml),并将该混合物用etoac萃取三次。将合并的有机溶液用盐水洗涤,并经mgso4干燥。使用10%的meoh在etoac中的溶液的制备型tlc产生10.4mg(34%)作为澄清膜样固体的期望产物化合物170。ms:[m+1]=447。
1396、实施例27:化合物52的合成:
1397、
1398、方案17.
1399、
1400、将起始醇43(160mg,0.45mmol)用三溴氧磷(phosphorous oxide tribromide)(400mg,1.4mmol)在乙腈(10ml)中的溶液在80℃处理5小时。然后将反应混合物冷却至0℃,用饱和的nahco3猝灭,并用二氯甲烷萃取两次。将合并的二氯甲烷溶液用盐水洗涤,并经mgso4干燥。过滤,并在真空中除去溶剂,得到173.3mg(92%)作为微黄色泡沫状固体的溴化物。ms:[m+1]=418。
1401、向溴化物(55mg,0.131mmol)在二甲氧基乙烷(2ml;脱气)中的悬浮液中加入2mna2co3(0.39ml,0.78mmol)和3-氯苯基硼酸(42.2mg,0.27mmol)。将该反应混合物在室温搅拌2分钟,然后加入pd(pph3)4(75mg,0.065mmol),并将该悬浮液在85℃油浴中加热90分钟。冷却后,将该反应混合物用etoac稀释,并用盐水洗涤。将水层分离,并用etoac萃取三次。将所有的有机层合并,并经na2so4干燥,然后过滤,并在真空中除去溶剂。将产物通过连续制备型tlc纯化分离,其中使用20%的己烷类在etoac中的溶液,随后使用5%的meoh在dcm中的溶液。得到9.6mg作为淡棕色固体的产物(化合物52)。ms:[m+1]=450。1h-nmr(500mhz,cdcl3)δ:7.75(s,1h),7.64(d,1h,j=3.0hz),7.51(d,1h,j=9.5hz),7.31(br s,1h),7.23(br s,1h),7.17(m,3h),4.43(q,2h,j=7.0hz),4.15(s,2h),3.96(s,3h),1.44(t,3h,j=8.0hz)。
1402、实施例28:化合物53的合成:
1403、
1404、以与关于实施例27描述的途径类似的合成途径,在最终的步骤中使用3-氰基苯基硼酸,合成实施例28的化合物,以得到作为淡棕色固体的实施例28的化合物:ms:[m+1]=441。1h-nmr(500mhz,cdcl3)δ:7.75(s,1h),7.66(br s,1h),7.64(d,1h,j=3.0hz),7.61(br d,1h,j=7.5hz),7.39(t,1h,j=7.5hz),7.16(dd,1h,j=3.5,9.5hz),4.45(q,2h,j=7.0h),4.20(s,2h),3.96(s,3h),1.45(t,3h,j=7.0hz)。
1405、实施例29:化合物54的合成:
1406、
1407、以与关于实施例27描述的途径类似的合成途径,从醇(其中r1=甲基)开始且在最终的步骤中使用2-氯苯基硼酸,合成实施例29的化合物,以得到作为淡棕色固体的实施例29的化合物:ms:[m+1]=434。
1408、实施例30:化合物101的合成:
1409、
1410、以与关于实施例27描述的途径类似的合成途径,从醇(其中r1=甲基)开始且在最终的步骤中使用苯基硼酸,合成实施例30的化合物,以得到作为淡棕色固体产物的实施例30的化合物,将其通过色谱法(redisep 4g硅胶柱。洗脱溶剂:etoac),然后通过制备型tlc(洗脱系统:40%dcm/40%己烷类/17%etoac/3%meoh)纯化以得到5.9mg(收率31%)产物化合物101。ms:[m+1]=402。h1nmr(cdcl3)δ7.96(1h,s),7.77(1h,s),7.55(1h,m),7.47(1h,m),7.32(5h,m),4.41(2h,q,j=7hz),4.17(2h,s),2.53(3h,s),1.43(3h,t,j=7hz)。
1411、实施例31:化合物102的合成:
1412、
1413、向所述溴化物在etoac(2ml)和meoh(2ml)中的悬浮液中加入活化的10%pd/c(5mg)。将该悬浮液在氢气气氛下搅拌48小时。将该溶液穿过硅藻土过滤。将滤液浓缩,并通过色谱法(redisep 4g硅胶柱。洗脱溶剂:etoac)纯化以得到15.9mg(33%)期望产物化合物102。ms:[m+1]=324。h1nmr(cdcl3)δ7.96(1h,s),7.78(1h,s),7.49(1h,d,j=9hz),7.42(1h,d,j=8hz),4.43(2h,q,j=7.5hz),2.53(3h,s),2.44(3h,s),1.45(3h,t,j=7.5hz)。
1414、实施例32:化合物108的合成:
1415、
1416、向溴化物衍生物(其中r1=ome)(18mg;0.043mmol)在除氧的dme(2ml)中的悬浮液中加入2-氯苯基硼酸(10mg,0.065mmol)和2mna2co3溶液(0.13ml,0.26mmol)。将该悬浮液在室温搅拌15分钟,然后加入pdcl2dppf(7mg,0.009mmol)。将该悬浮液在油浴中在85c加热1小时。将该反应混合物用水稀释,并用etoac(两次)萃取。将合并的萃取物用盐水洗涤,并经na2so4干燥。过滤并浓缩,得到粗产物,将其通过preptlc(洗脱系统:5%meoh/47.5%hex/47.5%etoac)纯化以得到3.5mg(收率18%)产物化合物108。ms:[m+1]=451。h1nmr(cdcl3)δ7.77(1h,s),7.63(1h,d,j=3hz),7.52(1h,d,j=11.5hz),7.36(1h,m),7.31(1h,m),7.18(2h,m),7.14(1h,dd,j=3,9hz),4.38(2h,q,j=7hz),4.27(2h,s),3.94(3h,s),1.41(3h,t,j=7hz)。
1417、方案18a.
1418、
1419、方案18b.
1420、
1421、实施例33:化合物55的合成:
1422、
1423、向化合物58(6.6g,33.5mmol)在二氯甲烷(100ml)中的溶液中加入dipea(8.65g,67mmol),hobt(5.4g,36.85mmol)和edci(9.6g,50.3mmol)。搅拌约15分钟后,在氮气气氛下向该均相反应混合物中逐滴加入2,4-二甲氧基苄基胺(5.6g,33.5mmol)在二氯甲烷(50ml)中的溶液。将得到的混合物在氮气气氛下在室温搅拌16h。将该反应混合物用1n naoh(100ml),水(100ml)和盐水(100ml)连续洗涤。然后将有机相经na2so4干燥,并蒸发,得到粗制固体产物59,将其从乙醚中结晶。过滤和敞口抽滤干燥,得到灰白色固体纯产物9.8g(96%),(ms:[m+1]=347)。
1424、向化合物59(9.8g,28.3mmol)在meoh/etoac(1:1,100ml)中的溶液中加入10%的湿的pd-c(1.8g,10%mmol)。连续三次抽真空和氮气净化后,将该非均相反应混合物在大气压下进行气球氢化,直至停止吸收氢气,约4h。将该反应混合物穿过硅藻土垫过滤,并蒸发,得到8.63g(96%)作为棕色油的纯期望产物60(ms:[m+1=317])。将该产物直接用于下一步。
1425、向化合物60(8.63g,27.3mmol)在二氯甲烷(100ml)中的溶液中加入三乙胺(5.5g,54.6mmol)。将该混合物用冰浴冷却,并在氮气气氛下用溴乙酰氯(5.2g,32.76mmol)处理。除去冰浴,并将该混合物放置搅拌18h。将该反应混合物用饱和的nahco3(100ml),水(100ml)和盐水(100ml)连续洗涤。然后将有机相经na2so4干燥,并蒸发,得到粗制固体产物61。将粗产物从甲醇中结晶,过滤,并干燥,得到棕色固体纯产物10.3g(87%),[ms:439]。
1426、向化合物61(10g,22.9mmol)在dmf(1000ml)中的溶液中加入k2co3(4.8g,45.8mmol)。将该混合物在50℃加热24h。lcms指示完全转化为期望产物。将该混合物冷却至室温,并将无机固体过滤。将溶剂在高真空下除去。将得到的粗产物62从甲醇中结晶,过滤,并干燥,得到6.4g(78%)纯棕色固体产物,(ms:[m+1]=357)。
1427、在-20℃向溶解在2.5:1thf/dmf(50ml)中的化合物62(4.46g,12.52mmol)中加入t-buok(97%,1.88g,16.28mmol)。将该混合物温热至25℃,并在搅拌30分钟后,再次冷却至-20℃。逐滴加入氯磷酸二乙酯(2.35ml,16.28mmol)后,将该混合物搅拌3小时,同时从-20℃温热至25℃。将该反应混合物再冷却至0℃,并向其中加入异氰基乙酸乙酯(1.92ml,17.53mmol)。然后冷却至-78℃,随后加入t-buok(97%,1.88g,16.28mmol)并在室温搅拌5小时。通过lc/ms监测过程。通过加入1:1饱和的nahco3/h2o(140ml)将该反应猝灭,将沉淀物过滤,用h2o洗涤并风干过夜,得到4.81g(85%)作为黄色固体的咪唑衍生物63(ms:[m+1]=452)。
1428、在0℃向在二氯甲烷(35ml)中的化合物63(4.81g,10.65mmol)中加入三氟乙酸(35ml),随后逐滴加入三氟甲磺酸(1.9ml,21.31mmol)。将该混合物温热至室温,搅拌2小时,然后浓缩,得到残余物,将其溶解在二氯甲烷(120ml)中。将该粗制溶液在冷却的饱和的nahco3和二氯甲烷之间分配。将有机萃取物合并,干燥(mgso4),过滤并浓缩,得到3.2g(99%)去保护的产物64(棕色固体),其纯度足够进行下一步(ms:[m+1]=302)。
1429、在氮气下向在氯苯(1ml)中搅拌的内酰胺64(51.8mg,0.172mmol)和n,n-二甲基-对甲苯胺(93.0mg,0.688mmol)中加入pocl3(52.7mg,0.344mmol)。然后将该反应物在135℃加热2小时。冷却至室温后,将苯氧基乙酸酰肼(228.4mg,1.36mmol)原位加入亚氨基-氯化物65中,随后加入dipea(90ul)。将该反应物在室温搅拌30分钟,然后在100℃加热90分钟。将该反应混合物冷却,加入饱和的nahco3(水溶液),并用乙酸乙酯萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物化合物55通过isco快速柱色谱法(redisep 4g柱,1-10%的meoh在dcm中的溶液作为洗脱梯度)分离为白色固体,重量:8.6mg。ms:[m+1]=432。1h-nmr(500mhz,cdcl3)δ:7.81(s,1h),7.71(d,1h,j=3.5hz),7.52(d,1h,j=9.0hz),7.32(m,2h),7.21(dd,1h,j=2.5,8.5hz),7.11(d,2h,j=8.5hz),7.02(m,1h),5.44(s,2h),4.38(q,2h,j=7.5hz),3.94(s,3h),1.39(t,3h,j=7.0hz)。
1430、实施例34:化合物56的合成:
1431、
1432、以与关于实施例33描述的途径类似的合成途径,在最终的步骤中使用4-氟-苯氧基乙酸酰肼,合成实施例34的化合物,以得到作为微黄色固体的实施例34的化合物:ms:[m+1]=450。1h-nmr(500mhz,cdcl3)δ:7.82(s,1h),7.73(d,1h,j=3.5hz),7.53(d,1h,j=10.0hz),7.22(dd,1h,j=3.5,9.0hz),7.08-6.99(m,4h),5.41(s,2h),4.41(q,2h,j=7.0hz),3.95(s,3h),1.42(t,3h,j=6.5hz)。
1433、实施例35:化合物103的合成:
1434、
1435、以与关于实施例33描述的途径类似的合成途径,在最终的步骤中使用2-甲氧基乙酸酰肼,合成实施例35的化合物,以得到实施例35的化合物作为微黄色固体:ms:[m+1]=370。
1436、实施例36:化合物118的合成:
1437、
1438、将乙酰胺肟(8.4mg,0.108mmol)在甲苯中在rotavap上共沸三次,然后悬浮于thf(1.0ml)中。加入nah(60%无机悬浮液;3.3mg,0.081mmol),并将该混合物在室温搅拌10分钟。接着加入酯55(23.2mg,0.054mmol)。在室温搅拌40min后,将该反应混合物在70℃加热4小时。冷却后,将冷水(5ml)加入该反应混合物中,并将沉淀物通过过滤收集,用水洗涤,并干燥,以得到9.7mg(41%)作为微黄色固体的期望产物。ms:[m+1]=442。
1439、实施例37:化合物128的合成:
1440、
1441、以与上面关于实施例36描述的途径类似的合成途径,在最终的步骤中使用酯化合物103,合成实施例37的化合物,以得到作为淡棕色固体的实施例37的化合物:ms:[m+1]=380。
1442、实施例38:化合物130的合成:
1443、
1444、以与关于实施例36描述的途径类似的合成途径,从酯化合物103开始并与异丁脒肟缩合,合成实施例38的化合物,以得到作为微黄色固体的实施例38的化合物:ms:[m+1]=408。
1445、实施例39:化合物119的合成:
1446、
1447、向在dcm(0.2ml)中搅拌的羧酸(13.9mg,0.0345mmol;通过前体酯55的lioh水解获得)中加入新戊醇(30.4mg,0.345mmol),dmap(4.2mg,0.0345mmol)和edc(20mg,0.104mmol)。搅拌5小时后,将该反应混合物用etoac稀释,用饱和nh4cl,饱和nahco3,盐水洗涤,并经mgso4干燥。使用0-8%的meoh在etoac中的溶液梯度的硅胶色谱纯化产生11.7mg(72%)作为微黄色固体的期望产物化合物119。ms:[m+1]=474。
1448、实施例40:化合物120的合成:
1449、
1450、以与上面关于实施例39描述的途径类似的合成途径,在最终的步骤中使用2-丙醇,合成实施例40的化合物,以得到作为微黄色固体的实施例40的化合物:ms:[m+1]=446。
1451、实施例41:化合物129的合成:
1452、
1453、通过用lioh(21.4mg,0.895mmol)在室温处理2小时,将化合物103(方案18a)(66.1mg,0.179mmol)在thf/水/meoh的溶剂系统(总计1.8ml,比例6/5/1)中水解。加入稀hcl以酸化(ph~3)该反应混合物。将沉淀物通过过滤收集,用水洗涤,并干燥,以得到49.0mg(80%)作为淡棕色固体的酸。
1454、在0℃将如此得到的酸在dmf(0.7ml)中搅拌。加入nahco3(48.1mg,0.572mmol),随后加入n-溴琥珀酰胺(96.7mg,0.543mmol)。搅拌过夜后,将该反应物用etoac稀释,并用饱和的nahco3洗涤。将水层分离,并用etoac萃取。将合并的有机层用盐水洗涤,经mgso4干燥,过滤并浓缩。通过使用0-13%的meoh在etoac中的溶液梯度洗脱的硅胶柱色谱法得到作为白色固体的产物溴化物(化合物129)。重量:28.6mg(53%)。ms:[m+1]=377。
1455、实施例42:化合物131的合成:
1456、
1457、将化合物129(22.6mg,0.060mmol)在10%pd-c上在etoac(1ml)和meoh(1ml)中氢化16小时。穿过硅藻土过滤,并除去溶剂,得到14.9mg(84%)作为轻微黄色固体的脱溴产物化合物131。ms:[m+1]=298。
1458、实施例43:化合物122的合成:
1459、
1460、将酸66(20.4mg,0.0506mmol)的苯氧基类似物(方案18a,r1=oph)在dcm(0.5ml)中在室温混悬和搅拌。加入羰基二咪唑(16.4mg,0.101mmol)。搅拌2h后,将得到的悬浮液冷却至0℃,并逐滴加入氨水(30ul)。搅拌20min后,除去冰浴,并使该反应在室温进行1小时。通过在真空中除去dcm,将该反应物浓缩。加入水(3ml),将沉淀物通过过滤收集,用水洗涤,并干燥,得到16.2mg粗制伯酰胺,将其不经进一步纯化地使用。
1461、将该伯酰胺(16.2mg,0.0402mmol)在95℃用pocl3(46.2mg,0.302mmol)在1,4-二噁烷(0.5ml)中的溶液处理过夜。然后将该反应混合物用饱和nahco3(5ml)猝灭,冷却至0℃,并将沉淀物通过抽滤收集,用水洗涤,并干燥,以得到13.6mg(88%)作为淡棕色固体的腈,化合物122。ms:[m+1]=385。
1462、实施例44:化合物123的合成:
1463、
1464、向在thf(0.15ml)和dcm(0.15ml)中搅拌的酸66(15.8mg,0.0392mmol)中加入n,o-二甲基羟胺hcl(4.6mg,0.047mmol)和n-羟基苯并三唑水合物(6.0mg)。然后加入edc(11.3mg,0.0588mmol)和三乙胺(11.9mg,0.118mmol),并将该反应混合物在室温搅拌12小时,用etoac稀释,用饱和的nh4cl,盐水洗涤,并经mgso4干燥。过滤,并在真空中除去溶剂,得到14.4mg(82%)weinreb酰胺,将其不经进一步纯化地使用。
1465、在0℃向在thf(0.3ml)中搅拌的weinreb酰胺(14.4mg,0.0323mmol)中加入乙基溴化镁乙醚络合物(3m;0.323ml)。使该反应物温热至室温并搅拌14小时,用饱和的nh4cl猝灭,用etoac萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制酮产物,将其通过使用8%的meoh在etoac中的溶液的制备型tlc纯化。重量:4.6mg(34%)化合物123。ms:[m+1]=416。
1466、实施例45:化合物124的合成:
1467、
1468、将上述的weinreb酰胺(18.0mg,0.0403mmol)用dibal(1m thf;0.363ml)在-78℃处理1小时,然后仍然在-78℃用罗谢尔盐溶液(20%)猝灭过夜。将该水溶液用etoac萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并在真空中除去溶剂,得到13.7mg的粗制醛,将其不经进一步纯化地使用。
1469、将在dcm(0.7ml)中的粗制醛(13.7mg)在室温用deoxo-fluor(54.8mg,0.248mmol)处理16小时。将该反应物用饱和的nahco3(5ml)猝灭20分钟,用etoac萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,随后通过使用10%的meoh在etoac中的溶液的制备型tlc纯化,得到7.5mg(52%)作为微黄色固体的期望的二氟化物化合物124。ms:[m+1]=410。
1470、实施例46:化合物142的合成:
1471、
1472、将在thf(0.15ml)中的上述weinreb酰胺(8.8mg,0.0197mmol)在0℃用苯基溴化镁(1m thf;0.54ml)处理2.5小时,用饱和的nh4cl猝灭,用etoac萃取两次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制酮,将其不经进一步纯化地使用。将在thf(0.5ml)中的该酮用nabh4(6mg)在室温处理2小时,然后用饱和的nh4cl猝灭,用etoac萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制醇,将其不经进一步纯化地使用。将在dcm(1.4ml)中的如此得到的醇用三乙基硅烷(86.4mg,0.75mmol)和三氟乙酸(171.0mg,1.5mmol)在40℃处理过夜,然后在真空中浓缩,用etoac稀释,用饱和的nahco3,盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制苄基产物,将其通过使用0-12%的meoh在etoac中的溶液作为洗脱液的硅胶柱色谱法纯化;得到3.6mg作为微黄色固体的化合物142。ms:[m+1]=450。
1473、方案19:
1474、
1475、实施例47:化合物106的合成:
1476、
1477、向在氯苯(5ml)中的内酰胺64(185.7mg,0.616mmol)中加入n,n-二甲基-对甲苯胺(333.3mg,2.465mmol)和磷酰氯(phosphorous oxychloride)(188.9mg,1.232mmol)。将该反应混合物在135℃加热2小时,冷却至室温,并加入甲酰基酰肼(296.0mg,4.93mmol),随后加入二异丙基乙基胺(238.8mg,1.85mmol)。在室温搅拌30分钟后,将该反应物在100℃加热1小时,冷却,并加入饱和nahco3(15ml),用etoac萃取两次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制三唑产物,将其通过使用0-15%的meoh在etoac中的溶液洗脱的硅胶柱色谱法纯化,得到35.9mg(18%)淡棕色固体。ms:[m+1]=326。
1478、在0℃将在dcm(1ml)中的来自上文的三唑用n-溴琥珀酰胺(37.6mg,0.21mmol)处理。使该反应物缓慢地温热至室温,并在室温反应过夜,用etoac稀释,用饱和的nahco3,盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制溴化物,将其通过使用0-10%的meoh在etoac中的溶液梯度的硅胶柱色谱法纯化;得到22.9mg(51%)作为灰白色固体的化合物106。[ms]:406。
1479、实施例48:化合物104的合成:
1480、
1481、给微波反应容器装入苯酚(20.3mg,0.216mmol),来自实施例47的溴化物底物(29.1mg,0.0719mmol),cs2co3(117.0mg,0.360mmol),1,3-丙酮二甲酸二乙酯(14.5mg,0.0719mmol)和dmf(0.5ml)。将该容器用氮气净化。加入cui(6.8mg,0.036mmol),并将该混合物在室温搅拌5min,随后在140℃在微波辐射条件下加热60分钟。将该反应混合物用etoac稀释,用水洗涤;将水层分离,并用etoac萃取两次;将合并的有机溶液用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗制醚产物,将其通过使用5%的meoh在dcm中的溶液的制备型tlc纯化;得到6.6mg作为微黄色固体的化合物104。ms:[m+1]=418。
1482、实施例49:化合物105的合成:
1483、
1484、以与上面关于实施例48描述的途径类似的合成途径,使用3-甲氧基苯酚替代苯酚,合成实施例49的化合物,以得到作为微黄色泡沫状固体的实施例49的化合物:ms:[m+1]=448。
1485、方案20:
1486、
1487、实施例50:化合物112的合成:
1488、
1489、向化合物2(160mg,0.49mmol)在thf(6ml),水(5ml)和meoh(1ml)中的溶液中加入lioh(59mg,2.45mmol)。将该溶液在室温搅拌3小时。将该溶液浓缩,并将粗制物质用1n hcl酸化直至ph 3-4。没有观察到固体。加入etoac,并将有机相(3×)萃取。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到112mg(77%收率)作为橙色固体的期望羧酸产物。ms:[m+1]=300。
1490、向酸(30mg,0.1mmol)在二氯乙烷(0.2ml)中的悬浮液中加入亚硫酰氯(0.4ml;5mmol)和dmf(20μl)。将得到的溶液在70c加热1小时。加入另外的0.2ml的亚硫酰氯,并将该溶液加热另外30分钟。将溶剂除去。将粗制物质在真空下干燥。
1491、将粗制的酰基氯(0.1mmol)悬浮于异丙醇中,并在室温搅拌18小时。将溶剂蒸发,并将粗制物质通过色谱法(redisep 4g硅胶柱,用10%的meoh在dcm中的溶液洗脱)纯化,得到8.6mg(25%收率)的产物化合物112[m+1]=342)。
1492、h1nmr(cdcl3)δ7.90(1h,d,j=9hz),7.79(1h,bs),7.63(1h,
1493、bs),7.36(1h,bs),3.48(1h,m),2.45(3h,s),1.43(6h,d,j=6.5hz)。
1494、实施例51:化合物113的合成:
1495、
1496、将上文制备的粗制酰基氯(0.066mmol)悬浮于二氯乙烷(1ml)中,并加入2,2-二甲基-1-丙醇(300mg,3.4mmol)。将该溶液在室温搅拌18小时。没有产物形成。向以上溶液中加入dmap(5mg,0.004mmol)和dcc(15mg,0.073mmol)。将该溶液在室温搅拌2小时。将该反应混合物直接应用于制备型tlc(洗脱系统:75etoac在己烷类中)以得到7.2mg(30%收率)的产物化合物113。ms:[m+1]=370。h1nmr(cdcl3)δ7.91(1h,dd,j=3,9hz),7.79(1h,s),7.61(1h,dd,j=4.5,9hz),7.35(1h,m),4.11(2h,s),2.44(3h,s),1.07(9h,s)。
1497、实施例52:化合物114的合成:
1498、
1499、将以上制备的粗制酰基氯(0.066mmol)悬浮于二氯乙烷(1ml)中,并加入2,2,2-三氟乙醇(0.1ml,1.4mmol),随后加入三乙胺(0.6ml,4.3mmol)。将该溶液在室温搅拌2小时30分钟。将溶剂蒸发,并将粗制物质通过色谱法(redisep 4g硅胶柱,用etoac洗脱)纯化,然后通过制备型tlc(洗脱系统:70%的etoac在己烷类中的溶液)纯化,得到8.1mg(32%收率)的产物化合物114[m+1]=382)。
1500、h1nmr(cdcl3)δ7.91(1h,dd,j=3.5,9.5hz),7.83(1h,s),7.63
1501、(1h,dd,j=4.5,9.5hz),7.35(1h,m),4.77(2h,m),2.43(3h,s)。
1502、实施例53:化合物136的合成:
1503、
1504、向用冰浴冷却的实施例50中制备的酸(100mg,0.33mmol)在dmf(1.5ml)中的溶液中加入nahco3(111mg,1.32mmol),随后加入nbs(117mg,0.66mmol)。将该溶液在室温搅拌14小时。将该反应混合物用水稀释,并用etoac(5x)萃取。将合并的萃取物用盐水(2×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱,用etoac洗脱)得到93mg(85%收率)的产物化合物136[m+1]=334)。h1nmr(cdcl3)δ7.87(1h,dd,j=2.5,8.5hz),7.72(1h,s),7.56(1h,dd,j=6,10hz),7.33(1h,m),2.44(3h,s)。
1505、实施例54.化合物139的合成:
1506、
1507、通用偶联程序:向化合物136(20mg,0.061mmol)在脱气的dme(0.9ml)和水(0.1ml)中的溶液中加入苯基硼酸(11mg,0.092mmol),碳酸铯(80mg,0.24mmol)和pd cl2dppf(5mg,0.066mmol)。将该悬浮液在80℃加热1小时。将该反应混合物用水稀释,用etoac(3x)萃取。将合并的萃取物用盐水(2×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物,将其通过制备型tlc(洗脱系统:3%的meoh在etoac中的溶液)纯化。
1508、使用苯基硼酸制备化合物139。得到10.8mg(54%收率)的产物。ms:[m+1]=332。h1nmr(cdcl3)δ7.87(1h,dd,j=3.5,9.5hz),7.85(1h,s),7.63(3h,m),7.50(2h,t,j=6.5hz),7.35(2h,m),2.41(3h,s)。
1509、实施例55:化合物140的合成:
1510、
1511、类似地使用3-吡啶硼酸制备化合物140。得到8.9mg(27%收率)的产物。ms:[m+1]=333。h1nmr(cdcl3)δ8.86(1h,s),8.63(1h,d,j=5hz),8.01(1h,m),7.90(2h,m),7.64(1h,dd,j=5.5,9hz),7.44(1h,m),7.36(1h,m),2.39(3h,s)。
1512、实施例56:化合物152的合成:
1513、
1514、使用1-甲基吡唑-4-硼酸,hcl,制备化合物152。得到12.5mg(63%收率)的产物。ms:[m+1]=336。h1nmr(cdcl3+meod4)δ9.04(1h,bs),7.99(1h,bs),7.75(2h,m),7.41(2h,m),3.95(3h,s),2.32(3h,s)。
1515、实施例57:化合物154的合成:
1516、
1517、使用2-甲基吡啶-4-硼酸频哪醇酯制备化合物154。得到7.1mg(34%收率)的产物。ms:[m+1]=347。h1nmr(cdcl3)δ8.6(1h,d,j=6hz),7.89(1h,dd,j=3.5,8.5hz),7.87(1h,s),7.64(1h,dd,j=5.5,9hz),7.48(1h,s),7.36(2h,m),2.64(3h,s),2.41(3h,s)。
1518、方案21:
1519、
1520、实施例58:化合物117的合成:
1521、
1522、在100ml圆底烧瓶中,将内酰胺酯16’(2g,7.35mmol;以与方案11中描述的16类似的方式制备)溶解在60ml的无水thf中。将该溶液在室温在氮气气氛下搅拌。缓慢地加入libh4(2m的在thf中的溶液,4ml,8mmol)。将该反应混合物在氮气气氛下搅拌18小时。缓慢地加入更多的libh4(2m的在thf中的溶液,2ml,4mmol)。将该反应混合物搅拌另外24小时。将etoac/etoh(20ml/20ml)的混合物加入该反应混合物中,并将其浓缩。将残余物溶解在meoh中,并加入硅胶。将挥发性溶剂蒸发后,将固体加载到redisep 40g硅胶柱上。将期望产物用5:1v/v ch2cl2/meoh。洗脱。得到作为白色固体的醇(1.14g,67%收率)。ms:[m+1]=231。
1523、将该醇(1.14g,4.96mmol)悬浮于16ml 33%的hbr在acoh中的溶液中,并在80℃加热18小时。将该溶液用冰浴冷却,并用etoac稀释。可以观察到白色固体。缓慢地加入饱和的nahco3水溶液。使用大量的etoac和meoh溶解固体。将有机相(3×)萃取,并将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到粗产物,将其不经进一步纯化地用于下一步。ms:[m+1]=293。
1524、向烷基溴衍生物(4.96mmol)在etoac(50ml),meoh(200ml)和thf(50ml)中的溶液中加入湿的10%pd/c(250mg),并将得到的悬浮液在氢气气氛下搅拌7天。将该悬浮液穿过硅藻土过滤,并将得到的溶液浓缩,并与甲苯共蒸发。将粗产物不经进一步纯化地用于下一步。
1525、在0℃向1,2,4-三唑(2.7g,39.7mmol)在无水ch3cn(20ml)中的溶液中加入i-pr2net(7.6ml,43.6mmol)。一旦所有三唑溶解,加入pocl3(1.11ml,11.9mmol)。将该混合物在0℃搅拌2小时。将该溶液转移进含内酰胺(4.96mmol)的烧瓶中。将得到的溶液在油浴中在80℃加热16小时。将该粘稠混合物用冰浴冷却,并蒸发溶剂。用etoac稀释,并加入水。将其用etoac萃取5次。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物,将其直接用于下一个反应中。ms:[m+1]=266。
1526、将kotbu(1.11g,9.92mmol)在dmf(10ml)中的溶液在氮气气氛下冷却至-50℃。缓慢地加入异氰基乙酸乙酯(1.2ml,10.9mmol)。将该混合物在-60℃至-40℃之间搅拌1小时。缓慢地加入在dmf(5ml)中的以上来自步骤4的粗制1,2,4-三唑并中间体(4.96mmol)。历时16h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 24g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到296mg(20%收率,4个步骤)产物。ms:[m+1]=310。
1527、向酯衍生物(260mg,0.84mmol)在thf(6ml),水(5ml)和meoh(1ml)中的溶液中加入lioh(117mg,4.85mmol)。将该溶液在室温搅拌3小时。将该溶液浓缩,并将粗制物质用1nhcl酸化直至ph 3-4。通过多重过滤收集固体,得到178mg(75%收率)的期望产物。ms:[m+1]=282。
1528、向酸(80mg,0.28mmol)在thf(2ml)中的悬浮液中加入cdi(50mg,0.31mmol)。将该悬浮液在65c加热3小时。lcms指示反应未完成。加入更多的cdi(10mg),并将溶液加热另外1小时。将该溶液冷却至室温,并加入nh4oh溶液(1ml)。将该溶液搅拌1小时。将固体通过过滤收集,得到33mg(42%)作为白色固体的期望产物化合物117。ms:[m+1]=281。h1nmr(meod4)δ8.1(1h,s),7.9(1h,s),7.73(3h,m),7.07(2h,s),2.40(3h,s)。
1529、实施例59:化合物115的合成:
1530、
1531、向化合物117(8mg,0.029mmol)和三乙胺(8μl;0.058mmol)在thf(1ml)中的悬浮液中加入三氟乙酸酐(8μl;0.058mmol)。将该反应混合物在室温搅拌16小时。lcms指示仅30%转化率。加入更多的三氟乙酸酐(30μl)和三乙胺(30μl)。该溶液变得澄清,搅拌另外1小时。将该反应用meoh猝灭。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:70%的etoac在己烷类中的溶液)纯化以得到6.6mg(83%)化合物115。ms:[m+1]=263。h1nmr(cdcl3)δ8.17(1h,d,j=7hz),7.88(1h,s),7.67(3h,m),2.46(3h,s)。
1532、实施例60:化合物127的合成:
1533、
1534、向化合物115(16mg,0.06mmol)在etoh(0.8ml)和水(0.2ml)中的悬浮液中加入盐酸羟胺(6mg,0.09mmol)和碳酸钾(12mg,0.09mmol)。将该悬浮液在80℃加热16小时。将该溶液用etoac稀释,并用水洗涤。将水层分离,并用etoac(3×)萃取。将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到12.2mg(67%收率)的期望产物。ms:[m+1]=296。
1535、将肟(10mg,0.034mmol)在乙酸酐(0.5ml)中的悬浮液在110c加热1小时。然后将该溶液在130c加热1小时。最后将温度升至140℃,并加热另外2小时。将该反应混合物冷却,并将etoh(1ml)加入该反应混合物中,将其在80℃加热16小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:etoac)纯化以得到6.1mg(56%收率)的期望产物化合物127。ms:[m+1]=320)。h1nmr(cdcl3)δ8.16(1h,m),7.92(1h,s),7.65(3h,m),2.68(3h,s),2.46(3h,s)。
1536、实施例61:化合物133的合成:
1537、
1538、向异丁酸(19μl,0.2mmol)在thf(0.5ml)中的溶液中加入cdi(10mg,0.062mmol)。将该溶液在室温搅拌2小时。然后将该溶液转移进含上文描述的肟衍生物(12mg,0.041mmol)的小瓶中,并在70℃加热2小时。lcms指示反应未完成。制备另一批次的试剂(异丁酸和cdi),并加入该反应混合物中,将其在70℃加热另外1小时。lcms指示所有起始原料被消耗。将溶剂蒸发,并将粗制物质悬浮于异丁酸(1ml)中,并在130℃加热1小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:70%的etoac在己烷类中的溶液)纯化以得到6.7mg(71%)期望产物化合物133。ms:[m+1]=348。
1539、h1nmr(cdcl3)δ8.16(1h,m),7.92(1h,s),7.65(3h,m),3.32(1h,m),2.46(3h,s),1.5(6h,d,j=7hz)。
1540、实施例62:化合物126的合成:
1541、
1542、将乙酰胺肟在使用前在甲苯中共沸三次。向乙酰胺肟(24mg,0.32mmol)在thf(1ml)中的悬浮液中加入60%的nah在油中的分散体(13mg,0.32mmol)。将该悬浮液在室温搅拌15分钟。加入化合物3(50mg,0.16mmol)。将含酯的小瓶用加入该反应混合物中的dmf(1ml)漂洗。将得到的棕色悬浮液在室温搅拌30分钟,然后在70℃加热2小时。将该悬浮液用水猝灭,并将该溶液保存在冰箱内过夜。通过多重过滤收集固体,得到16mg(31%收率)的产物化合物126。ms:[m+1]=320。h1nmr(cdcl3)δ8.18(1h,m),7.94(1h,s),7.67(3h,m),2.51(3h,s),2.46(3h,s)。
1543、实施例63:化合物125的合成:
1544、
1545、向衍生自化合物3的羧酸(30mg,0.11mmol),n,o-二甲氧基胺盐酸盐(13mg,0.13mmol),1-羟基苯并三唑水合物(17mg,0.11mmol)和三乙胺(46μl,0.33mmol)在thf(0.3ml)和dcm(0.3ml)中的悬浮液中加入1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(32mg,0.17mmol)。将该溶液在室温搅拌16小时。将该反应混合物用饱和的氯化铵溶液猝灭,并用etoac(3×)萃取。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到31.2mg(88%收率)的橙色固体,将其不经进一步纯化地用于下一步。ms:[m+1]=325。
1546、向在-78℃冷却的以上的weinreb酰胺衍生物(31.2mg,0.093mmol)在thf(0.5ml)中的溶液中加入3m乙基溴化镁的溶液(0.31ml,0.93mmol)。将该反应混合物在-10℃以下搅拌60分钟的时间。然后将其用饱和的氯化铵溶液猝灭,并用etoac(2×)萃取。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱,用80%的etoac在己烷类中的溶液洗脱)得到11.1mg(41%收率)的产物化合物125。ms:[m+1]=294。h1nmr(cdcl3)δ8.15(1h,m),7.76(1h,s),7.65(3h,m),3.08(2h,q,j=7hz),2.44(3h,s),1.22(3h,t,j=7hz)。
1547、实施例64:化合物132的合成:
1548、
1549、向异丁腈(2.6ml;29mmol)在etoh(30ml)和水(10ml)中的溶液中加入盐酸羟胺(2.01g,29mmol)和碳酸钾(4g,29mmol)。将得到的悬浮液在80℃加热16小时。将溶剂在真空下除去。将残余物与甲苯一起共蒸发。将粗制物质用etoh洗涤并过滤以除去氯化钠。将滤液蒸发,与甲苯共蒸发数次,并在真空下干燥,得到2g(69%)n-羟基丁脒。
1550、向n-羟基丁脒(47mg,0.46mmol)在thf(1ml)中的悬浮液中加入60%的nah在油中的分散体(18mg,0.46mmol)。将该悬浮液在室温搅拌30分钟。加入化合物3(47mg,0.15mmol)在thf(1ml)中的溶液。将得到的悬浮液在室温搅拌30分钟,然后在70℃加热2小时。1小时后,观察到仅50%转化率。另外1小时后没有观察到变化。制备如上文所述的更多的试剂(n-羟基丁脒和nah),并加入反应混合物中,将其加热另外40分钟。这时,lcms指示该反应完成。将该悬浮液用水猝灭。加入一些meoh以帮助完成溶解,并将该溶液用etoac(3×)萃取。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep4g硅胶柱,用etoac洗脱)得到20mg(38%收率)的产物化合物132。ms:[m+1]=348。h1nmr(cdcl3)δ8.18(1h,d,j=8hz),7.93(1h,s),7.69(3h,m),3.22(1h,m),2.46(3h,s),1.43(6h,d,j=9.5hz)
1551、实施例65:化合物161的合成:
1552、
1553、向用冰浴冷却的衍生自化合物3的酸(90mg,0.32mmol)在dmf(2ml)中的溶液中加入nahco3(108mg,1.28mmol),随后加入nbs(114mg,0.64mmol)。将该溶液在室温搅拌18小时。将该反应混合物用水稀释,并用etoac(3×)萃取。将合并的萃取物用盐水(2×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱,用etoac洗脱)得到54mg(53%收率)的产物。ms:[m+1]=316。
1554、向溴化物衍生物(30mg,0.1mmol)在二噁烷(1ml)和三乙胺(1ml)中的溶液中加入tms-乙炔(71μl,0.5mmol),cui(2mg,0.01mmol)和pdcl2(pph3)2(7mg,0.01mmol)。将该溶液在110℃加热6小时。加入更多的pd催化剂(7mg)和tms-乙炔(0.2ml),并将该反应混合物加热另外12小时。这时,lcms指示约80%转化率。加入更多的pd催化剂(7mg)和tms-乙炔(0.2ml),并将该反应混合物加热另外12小时。lcms指示完全转化。然后将该反应混合物用水稀释,并用etoac(3×)萃取。将合并的萃取物用盐水(2×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到23mg(69%收率)的产物。ms:[m+1]=334。
1555、在0℃向炔烃衍生物(23mg,0.069mmol)在meoh(0.6ml)和h2o(0.2ml)中的溶液中加入koh(4mg,0.076mmol)。历时16h将该溶液温热至室温。将该反应混合物用饱和的氯化铵水溶液稀释,并用etoac(2×)萃取。将合并的萃取物用盐水(2×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物,将其通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到8.1mg(45%收率)的产物化合物161。ms:[m+1]=262。h1nmr(cdcl3)δ8.13(1h,m),7.76(1h,s),7.62(3h,m),4.09(2h,bs),3.28(1h,s),2.44(3h,s)。
1556、实施例66:化合物146的合成:
1557、
1558、向3-氨基-2-甲基丙烯醛(65mg,0.76mmol)在无水thf(2ml)中的溶液中加入60%的nah在油中的分散体(30mg,0.76mmol)。将该悬浮液在室温搅拌15分钟。加入化合物115(50mg,0.19mmol),并将该反应混合物在65℃加热3小时。将该反应混合物用冰浴冷却,并加入水。将该反应混合物在冰箱中保存过夜。将固体通过过滤收集,得到27.5mg(44%收率)的白色固体化合物146。ms:[m+1]=330。h1nmr(cdcl3)δ8.66(2h,s),8.15(1h,m),7.89(1h,s),7.65(3h,m),2.44(3h,s),2.36(3h,s)。
1559、实施例67:化合物153的合成:
1560、
1561、向衍生自化合物3的酸(30mg,0.11mmol)在二氯乙烷(0.2ml)中的悬浮液中加入亚硫酰氯(1ml;13.8mmol)和dmf(20μl)。将得到的溶液在70℃加热1小时。将溶剂除去。将粗制物质在真空下干燥。将粗制物质悬浮于异丙醇(2ml)中,并在室温搅拌16小时。将溶剂蒸发,与甲醇一起共蒸发,并将粗制物质通过制备型tlc(洗脱系统:etoac)纯化以得到7.2mg(21%收率)的产物化合物153。ms:[m+1]=324。h1 nmr(cdcl3)δ8.15(1h,d,j=8hz),7.81(1h,s),7.64(3h,m),5.32(1h,q,j=7hz),2.45(3h,s),1.43(6h,d,j=7hz)。
1562、方案22
1563、
1564、实施例68:化合物116的合成:
1565、
1566、还实现了腈-取代的咪唑衍生物的一个供选择的合成途径。作为一个例子,如在方案22中所示,从亚氨基-衍生物制备化合物116。将异氰基乙腈(206mg,3.12mmol)在dmf(7ml)中的溶液在氮气气氛下冷却至-50℃。加入kotbu(320mg,2.85mmol)。将该混合物在-50℃搅拌1小时。在-50℃缓慢地加入亚氨基衍生物(以与上文方案21中所示的亚氨基衍生物相同的方式制备)(350mg,1.24mmol)。历时16h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 12g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到230mg(70%收率)的产物化合物116。ms:[m+1]=281。h1nmr(cdcl3)δ7.92(1h,dd,j=3,8.5hz),7.81(1h,s),7.61(1h,dd,j=4.5,9hz),7.38(1h,m),2.47(3h,s)。
1567、实施例69:化合物145的合成:
1568、
1569、向氰化物衍生物化合物116(50mg,0.18mmol)在etoh(1.6ml)和水(0.4ml)中的悬浮液中加入盐酸羟胺(17mg,0.24mmol)和碳酸钾(28mg,0.2mmol)。将该悬浮液在80℃加热30分钟,然后冷却至室温。通过过滤收集固体沉淀物,得到37.8mg(68%收率)的期望的氨基肟产物,[m+1]=314。
1570、将酰胺肟(10mg,0.032mmol)在乙酸酐(0.5ml)中的悬浮液在140c加热4小时。将该反应混合物冷却,并将etoh(1ml)加入该反应混合物中,将其在80℃加热16小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:etoac)纯化以得到6.6mg(61%收率)的期望产物化合物145。ms:[m+1]=338。h1nmr(cdcl3)δ7.91(1h,dd,j=3.5,8.5hz),7.89(1h,s),7.65(1h,dd,j=5.5,10hz),7.35(1h,m),2.69(3h,s),2.45(3h,s)。
1571、实施例70:化合物149的合成:
1572、
1573、向异丁酸(30μl,0.32mmol)在thf(0.5ml)中的溶液中加入cdi(16mg,0.096mmol)。将该溶液在室温搅拌2小时。加入以上酰胺肟衍生物(10mg,0.032mmol),并将该反应混合物在70c加热45分钟。将溶剂蒸发,并将粗制物质悬浮于异丁酸(1ml)中,并在130℃加热3小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到10.6mg(91%)期望产物化合物149。ms:[m+1]=366。h1nmr(cdcl3)δ7.90(1h,dd,j=3.5,9hz),7.89(1h,s),7.66(1h,dd,j=4.5,8.5hz),7.36(1h,m),3.32(1h,q,j=6.5hz),2.46(3h,s),1.49(6h,d,j=8hz)。
1574、实施例71:化合物150的合成:
1575、
1576、将以上酰胺肟(10mg,0.032mmol)在三氟乙酸酐(0.5ml)中的悬浮液在回流下加热10分钟。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到11.8mg(94%)期望产物化合物150。ms:[m+1]=392。h1nmr(cdcl3)δ7.92(2h,m),7.69(1h,dd,j=5.5,9.5hz),7.39(1h,m),2.45(3h,s)。
1577、实施例72:化合物151的合成:
1578、
1579、向甲酸(12μl,0.32mmol)在thf(0.5ml)中的溶液中加入cdi(16mg,0.096mmol)。将该溶液在室温搅拌2小时。加入以上酰胺肟衍生物(10mg,0.032mmol),并将该反应混合物在70℃加热45分钟。将溶剂蒸发,并将粗制物质悬浮于甲酸(1ml)中,并在60℃加热3小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到2.1mg(20%)期望产物化合物151。ms:[m+1]=324。h1nmr(cdcl3)δ8.83(1h,s),7.92(1h,dd,j=3.5,8hz),7.91(1h,s),7.65(1h,dd,j=4.5,9hz),7.37(1h,m),2.45(3h,s)。
1580、实施例73:化合物155的合成:
1581、
1582、向丙酸(22μl,0.29mmol)在thf(0.5ml)中的溶液中加入cdi(14mg,0.087mmol)。将该溶液在室温搅拌1小时。加入在thf(0.5ml)中的以上酰胺肟衍生物(10mg,0.032mmol),并将该反应混合物在70℃加热90分钟。将溶剂蒸发,并将粗制物质悬浮于丙酸(1ml)中,并在130℃加热1小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到9.4mg(94%)期望产物化合物155。ms:[m+1]=352。h1nmr(cdcl3)δ7.91(1h,dd,j=2,8.5hz),7.88(1h,s),7.65(1h,dd,j=6,9.5hz),7.36(1h,m),3.01(2h,q,j=8.5hz),2.46(3h,s),1.48(3h,t,j=8.5hz)。
1583、实施例74:化合物160的合成:
1584、
1585、向新戊酸(30mg,0.29mmol)在thf(0.5ml)中的溶液中加入cdi(14mg,0.087mmol)。将该溶液在室温搅拌1小时。加入在thf(0.5ml)中的以上酰胺肟衍生物(10mg,0.032mmol),并将该反应混合物在70℃加热90分钟。将溶剂蒸发,并将粗制物质悬浮于乙酸(1ml)中,并在回流下加热3小时。将溶剂蒸发,并将粗制物质通过制备型tlc(洗脱系统:80%的etoac在己烷类中的溶液)纯化以得到7.4mg(67%)期望产物化合物160。ms:[m+1]=380。h1nmr(cdcl3)δ7.90(1h,dd,j=2.7,9hz),7.88(1h,s),7.65(1h,dd,j=4.5,9hz),7.35(1h,m),2.47(3h,s),1.53(9h,s)。
1586、实施例75:化合物143的合成:
1587、
1588、将kotbu(40mg,0.36mmol)在dmf(3ml)中的溶液在氮气气氛下冷却至-50℃。加入对甲苯磺酰甲基异氰化物(76mg,0.39mmol)。将该混合物在-50℃搅拌1小时。加入来自方案22的亚氨基-衍生物(50mg,0.18mmol),并历时16h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取5次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 4g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)和随后的制备型tlc(洗脱系统:30%的etoac在dcm中的溶液)得到22.2mg(30%收率)的白色固体化合物143。ms:[m+1]=410。
1589、h1nmr(cdcl3)δ7.91(2h,d,j=8hz),7.87(1h,dd,j=2.5,8.5
1590、hz),7.74(1h,s),7.65(1h,dd,j=5.5,9hz),7.34(3h,m),2.50(3h,s),2.42(3h,s)。
1591、实施例76:化合物144的合成:
1592、
1593、向3-乙氧基异丁烯醛(100mg,0.88mmol)中加入7n的氨在甲醇中的溶液(1.3ml,8.8mmol)。将该溶液在室温搅拌16小时。将溶剂蒸发,并将与3-氨基-2-甲基丙烯醛对应的粗制黄色固体不经进一步纯化地用于下一步。
1594、向3-氨基-2-甲基丙烯醛(7mg,0.087mmol)在无水thf(1ml)中的溶液中加入60%的nah在油中的分散体(6mg,0.16mmol)。将该悬浮液在室温搅拌15分钟。加入在thf(1ml)中的氰化物衍生物(22mg,0.079mmol),并将该反应混合物在65℃加热1小时。如上文所述,用3-氨基-2-甲基丙烯醛(20mg)和nah(20mg)在thf(1ml)中的溶液制备一批新的试剂,并加入该反应混合物中,将其在65℃加热另外1小时。lcms指示反应完成。将该反应混合物用甲醇猝灭。将溶剂蒸发。将粗制物质悬浮于水中,并将固体通过过滤收集,得到5.2mg(19%收率)浅红色固体化合物144。ms:[m+1]=348。h1nmr(cdcl3)δ8.67(2h,s),7.90(1h,d,j=9.5hz),7.85(1h,s),7.65(1h,dd,j=4.5,9hz),7.34(1h,m),2.44(3h,s),2.36(3h,s)。
1595、方案23
1596、
1597、实施例77:化合物121的合成:
1598、
1599、在0℃向1,2,4-三唑(2.03g,29.4mmol)在无水ch3cn(20ml)中的溶液中加入i-pr2net(5.6ml,32.4mmol)。一旦所有三唑被溶解,加入pocl3(0.82ml,8.8mmol)和化合物16’(1g,3.68mmol)。将该混合物在0℃搅拌2小时。将得到的溶液在油浴中在80℃加热16小时。将该混合物用冰浴冷却,用etoac稀释,并加入水。将其用etoac萃取三次。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到1.05g(88%收率)的橙色固体,将其直接用在下一步中。ms:[m+1]=324。
1600、将kotbu(696mg,6.2mmol)在dmf(15ml)中的溶液在氮气气氛下冷却至-50℃。缓慢地加入异氰基乙酸乙酯(0.75ml,6.8mmol)。将该混合物在-50℃搅拌1小时。加入以上来自步骤1的粗产物(1g,3.1mmol),并历时18h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取8次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 24g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到950mg(83%收率)的产物。ms:[m+1]=368。
1601、在室温向在氮气气氛下搅拌的二酯(200mg,0.54mmol)在无水thf(4ml)中的溶液中加入libh4(2m的在thf中的溶液,0.66ml,1.3mmol)。将该反应混合物在氮气气氛下搅拌24小时。将etoac/etoh(3ml/3ml)的混合物加入该反应混合物中,并将其浓缩。将残余物溶解在meoh中,并加入硅胶。将挥发性溶剂蒸发后,将固体加载到redisep 4g硅胶柱上。将期望产物用10:1v/v ch2cl2/meoh洗脱。得到作为固体的二醇(60mg,39%收率)。ms:[m+1]=284。
1602、将该二醇(60mg,0.21mmol)悬浮于5ml 33%的hbr在acoh中的溶液中,并在80℃加热18小时。将该溶液用冰浴冷却,并用etoac稀释。缓慢地加入饱和的nahco3水溶液。将该溶液用etoac(3×)萃取,并将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到粗产物,将其不经进一步纯化地用于下一步。ms:[m+1]=408。
1603、向二烷基溴衍生物(0.21mmol)在etoac(10ml)和meoh(10ml)中的溶液中加入湿的10%pd/c(催化量),并将得到的悬浮液在氢气气氛下搅拌60小时。将该悬浮液穿过硅藻土过滤,并将得到的溶液浓缩。将粗产物通过多重制备型tlc(洗脱系统:3%的meoh在etoac中的溶液)纯化以得到6.2mg(12%收率,2步)的期望产物化合物121。ms:[m+1]=252。h1nmr(cdcl3)δ8.09(1h,m),7.74(1h,s),7.56(3h,m),7.90(2h,m),2.42(3h,s),2.29(3h,s)。
1604、实施例78:化合物135的合成:
1605、
1606、以与化合物121类似的方式如下合成化合物135:在0℃向1,2,4-三唑(952mg,13.8mmol)在无水ch3cn(20ml)中的溶液中加入i-pr2net(2.6ml,15.2mmol)。一旦所有三唑被溶解,加入pocl3(0.45ml,4.8mmol)和内酰胺酯(1g,3.45mmol)。将该混合物在0℃搅拌2小时。将得到的溶液在油浴中在80℃加热16小时。将该混合物用冰浴冷却,用etoac稀释,并加入水。将其用etoac萃取三次。将合并的萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到1.03g(87%收率)的橙色固体,将其直接用在下一步中。ms:[m+1]=342。
1607、将kotbu(658mg,5.9mmol)在dmf(15ml)中的溶液在氮气气氛下冷却至-50℃。缓慢地加入异氰基乙酸乙酯(0.71ml,6.5mmol)。将该混合物在-50℃搅拌1小时。加入以上步骤1的粗产物(1g,2.9mmol),并历时18h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取8次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep 24g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到1.02g(90%收率)的产物。ms:[m+1]=386。
1608、在室温向在氮气气氛下搅拌的二酯(600mg,1.56mmol)在无水thf(8ml)中的溶液中加入libh4(2m的在thf中的溶液,3.1ml,6.24mmol)。将该反应混合物在氮气气氛下搅拌24小时。将etoac/etoh(10ml/10ml)的混合物加入该反应混合物中,并将其浓缩。将残余物溶解在meoh中,并加入硅胶。将挥发性溶剂蒸发后,将固体加载到redisep12g硅胶柱上。将期望产物用10:1v/v ch2cl2/meoh洗脱。得到作为固体的二醇(187mg,40%收率)。ms:[m+1]=302。
1609、将该二醇(80mg,0.27mmol)悬浮于7ml的33%的hbr在acoh中的溶液中,并在80℃加热48小时。将该溶液用冰浴冷却,并用etoac稀释。缓慢地加入饱和的nahco3水溶液。将该溶液萃取(3×),并将合并的有机相用盐水洗涤,经mgso4干燥。过滤,浓缩,并与甲苯共蒸发,得到100mg(88%收率)的米色固体,将其不经进一步纯化地用于下一步。ms:[m+1]=426。
1610、向二烷基溴衍生物(70mg,0.16mmol)在etoac(10ml)和meoh(10ml)中的溶液中加入10%pd/c(催化量),并将得到的悬浮液在氢气气氛下搅拌48小时。将该悬浮液穿过硅藻土过滤,并将得到的溶液浓缩。将粗产物通过多重制备型tlc(洗脱系统1:75%的etoac在己烷类中的溶液;洗脱系统2:5%的meoh在etoac中的溶液;洗脱系统3:etoac)纯化以得到4.1mg(10%收率)的期望产物化合物135。ms:[m+1]=270。h1nmr(cdcl3)δ7.84(1h,dd,j=2.5,9hz),7.70(1h,s),7.54(1h,dd,j=5,8hz),7.30(1h,m),2.42(3h,s),2.28(3h,s)。
1611、实施例79:化合物134的合成:
1612、
1613、向在80℃加热的,在方案23中描述的二烷基溴衍生物(r=h)(30mg,0.074mmol)在etoh(1ml)中的悬浮液中加入新制备的2m的naoet溶液(75μl,0.15mmol)。将该溶液加热10分钟。将溶剂蒸发。将粗制物质悬浮于etoac中,并过滤。将滤液浓缩并通过制备型tlc(洗脱系统:etoac)纯化以得到3.1mg(12%收率)的期望产物化合物134。ms:[m+1]=340。
1614、方案24
1615、
1616、实施例80:化合物137的合成:
1617、
1618、向5-氟-2-硝基苯甲酸(6.6g,35.66mmol)在二氯甲烷(100ml)中的溶液中加入dipea(9.22g,71.3mmol),hobt(6.0g,39.2mmol)和edci(10.2g,53.5mmol)。搅拌约15分钟后,在氮气气氛下向该反应混合物中逐滴加入2,4-二甲氧基苄基胺(5.96g,35.66mmol)在二氯甲烷(50ml)中的溶液。将得到的混合物在氮气气氛下在室温搅拌16小时。将该反应混合物用1n hcl(100ml),饱和nahco3(100ml)和盐水(100ml)连续洗涤。然后将有机相经mgso4干燥。过滤,并在真空中除去溶剂,得到微黄色固体,重量:9.3g(78%)。ms:[m+1]=335。
1619、在室温向在hoac/thf/meoh/h2o(25/100/50/25ml)的溶剂混合物中混悬并搅拌的硝基苯类似物(9.3g,27.8mmol)中加入zn粉。将该混合物加热至70℃保持20小时,冷却,并过滤。将固体用thf漂洗,并将合并的滤液在真空中浓缩。向得到的浆料中缓慢地和小心地加入饱和的nahco3以避免过度形成,直至ph达到7至8。将该混合物用etoac(3×)萃取;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到作为深棕色树胶状糊的粗制胺产物,重量:8.7g。
1620、向以上的苯胺(8.7g)在二氯甲烷(150ml)中的溶液中加入三乙胺(3.37g,33.4mmol)。将该混合物用冰浴冷却,并在氮气气氛下用溴乙酰氯(4.81g,30.6mmol)处理。除去冰浴,将该混合物搅拌72小时。将该反应混合物在真空中浓缩,用et2o(100ml)和水(100ml)处理得到的浆料。将产物沉淀通过过滤收集,并干燥,得到5.6g作为棕色固体的产物。将et2o层与水层分离,并用dcm(50ml)稀释,用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到5.3g作为泡沫状棕色固体的另外产物。总重量:11g(100%)。
1621、向溴化物(11g)在dmf(550ml)中的溶液中加入k2co3(7.1g,51.7mmol)。将该混合物在50℃加热48小时。将该混合物冷却至室温,并将无机固体的滤出。将滤液在真空中浓缩,用水/meoh(60/10ml)处理,用dcm(3×)萃取;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,随后通过使用5-50%的etoac在dcm中的溶液的硅胶柱色谱法得到3.2g(36%)作为淡棕色固体的7-元内酰胺。ms:[m+1]=345。
1622、在-20℃向在thf(20ml)和dmf(3ml)中溶解并搅拌的内酰胺(1.32g,3.83mmol)中加入t-buok(0.645g,5.75mmol)。在-20℃搅拌30min后,逐滴加入氯磷酸二乙酯(1.19ml,6.89mmol),并将该混合物搅拌3小时,同时从-20℃温热至20℃。将该反应混合物冷却至-78℃,向其中加入异氰基乙酸乙酯(0.791ml,6.89mmol),随后加入t-buok(0.645g,5.75mmol),并继续搅拌过夜,同时使温度达到室温。将该反应用饱和的nh4cl猝灭,用etoac(2×)萃取;将合并的有机溶液用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到粗产物,将其通过使用15-100%的etoac在dcm中的溶液的硅胶柱色谱法纯化,重量:0.861g(47%),作为棕色固体。ms:[m+1]=440。
1623、在0℃向在二氯甲烷(5ml)中的上述咪唑酯(861mg)中加入三氟乙酸(5ml),随后加入三氟甲磺酸(0.345ml)。将该混合物温热至室温,搅拌3小时,然后浓缩得到残余物,将其溶解在二氯甲烷(50ml)中。向其中加入饱和的nahco3(50ml),随后搅拌20min。顶部水层的ph经测试为碱性,并将其分离,用dcm(3×)萃取;将合并的dcm溶液用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到0.58g(100%)作为微黄色固体的内酰胺。ms:[m+1]=290。
1624、在氮气下向在氯苯(2.5ml)中搅拌的内酰胺(209.1mg,0.723mmol)和n,n-二甲基-对甲苯胺(234.7mg,1.74mmol)中加入pocl3(133.0mg,0.867mmol)。然后将该反应混合物在135℃加热2小时。冷却至室温后,加入苯氧基乙酸酰肼(189.0mg,1.08mmol),随后加入dipea(0.455ml)。将该反应混合物在室温搅拌30分钟,然后在100℃加热60分钟。将该反应混合物冷却,加入饱和的nh4cl(水溶液),并用乙酸乙酯萃取三次;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物通过使用0-10%的meoh在etoac中的溶液的isco快速柱色谱法纯化,重量:116.7mg(36%)作为微黄色膜状固体的化合物137。ms:[m+1]=420。
1625、实施例81:化合物156的合成:
1626、
1627、将在thf/水/meoh的溶剂系统(总计6.0ml,比例6/5/1)中的乙酯化合物137(244.2mg,0.582mmol)用lioh(69.7mg,2.91mmol)在室温处理4小时,在真空中浓缩,酸化至ph~3,并将沉淀物通过过滤收集。水洗并干燥后,得到179.3mg(79%)的作为微黄色固体的酸。ms:[m+1]=392。
1628、在室温向在dcm(0.1ml)中搅拌的该酸(10.8mg,0.0276mmol)中加入edci(21.3mg,0.11mmol),dmap(6.7mg,0.0552mmol)和异丙醇(13.2mg,0.221mmol)。12小时后,将该反应物用etoac稀释,用饱和的nahco3洗涤;将水层分离,并用etoac萃取,将合并的有机层用盐水洗涤,并经mgso4干燥。将浓缩物过滤,并通过使用10%的meoh在etoac中的溶液的制备型tlc纯化,得到8.7mg(73%)作为微黄色泡沫状固体的异丙基酯化合物156。ms:[m+1]=434。
1629、实施例82:化合物138的合成:
1630、
1631、将乙酰胺肟(10.7mg,0.144mmol)在甲苯中共沸四次,并加入乙酯化合物137(9.5mg,0.0226mmol)中。加入thf(0.3ml),随后加入60%的nah油悬浮液(4.5mg,0.112mmol)。将该反应混合物在室温搅拌30分钟,然后在70℃加热2小时,冷却至室温,并在真空中除去溶剂,将水(1.5ml)加入以猝灭该反应,搅拌20分钟,并冷却至4℃。将沉淀物通过过滤收集,用水洗涤,并干燥,以得到5.2mg(59%)作为浅黄色固体的噁二唑产物化合物138。ms:[m+1]=430。
1632、实施例83:化合物141的合成:
1633、
1634、以与关于实施例82描述的途径类似的合成途径,使用异丁脒肟替代乙酰胺肟,合成实施例83的化合物,以得到作为微黄色固体的实施例83的化合物:ms:[m+1]=458。
1635、实施例84:化合物157的合成:
1636、
1637、在室温向在dcm(0.7ml)中搅拌的上述在实施例81中制备的酸(60.2mg,0.154mmol)中加入羰基二咪唑(49.9mg,0.308mmol)。将该混合物搅拌40分钟,然后冷却至0℃,并加入氨(0.112ml),温热至室温,同时持续搅拌过夜。将反应物浓缩,加入水(8ml),并充分搅拌30分钟。将得到的沉淀物通过过滤收集,用水洗涤,并干燥,得到51.1mg(85%)作为淡棕色固体的伯酰胺。ms:[m+1]=391。
1638、将来自上面的酰胺(51.1mg)用pocl3(200.8mg,1.31mmol)在1,4-二噁烷(0.9ml)中的溶液在90℃处理14小时。一旦冷却至室温,将该反应小心地用饱和的nahco3(5ml)猝灭,搅拌20分钟。将沉淀物通过过滤收集,用水洗涤,并干燥,得到40.9mg(85%)作为淡棕色固体的腈产物化合物157。ms:[m+1]=373。
1639、实施例85:化合物147的合成:
1640、
1641、向在圆底烧瓶中的腈(45.8mg,0.123mmol)中加入盐酸羟胺(14.5mg,0.209mmol),k2co3(22.3mg,0.161mmol),乙醇(0.6ml)和水(0.15ml)。将该反应混合物在80℃加热30min,冷却并在真空中浓缩。将得到的浆料用水(1.5ml)处理,声处理以帮助混合,并在室温搅拌1小时,然后冷却至4℃。将得到的沉淀物通过过滤收集,用冷水(1ml)洗涤,并干燥,得到40.8mg(82%)作为灰白色固体的加成化合物。ms:[m+1]=406。
1642、将异丁酸(31.4mg,0.582mmol)用羰基二咪唑(28.4mg,0.175mmol)在thf(0.5ml)中的溶液处理2小时。加入n-羟基甲酰胺加成化合物(11.8mg,0.0291mmol),并将该反应物在室温搅拌30分钟。加入更多的异丁酸(0.5ml),并将该反应混合物在110℃加热16小时,冷却,加入饱和的nahco3(8ml),并用etoac(3×)萃取;将合并的有机层用盐水洗涤,并经mgso4干燥。浓缩的滤液的制备型tlc(5%的meoh在etoac中的溶液)得到11.2mg(84%)作为白色固体的噁二唑化合物147。ms:[m+1]=458。
1643、实施例86:化合物148的合成:
1644、
1645、以与关于实施例85描述的途径类似的合成途径,使用乙酸替代异丁酸,合成实施例86的化合物,以得到作为白色固体的实施例86的化合物:ms:[m+1]=430。
1646、实施例87:化合物158的合成:
1647、
1648、以与关于实施例85描述的途径类似的合成途径,使用丙酸替代异丁酸,合成实施例87的化合物,以得到作为白色固体的实施例87的化合物:ms:[m+1]=444。
1649、实施例88:化合物159的合成:
1650、
1651、在室温将三氟乙酸酐(196.9mg,0.938mmol)加入在thf(0.2ml)中混悬并搅拌的n-羟基甲酰胺加成化合物(19.0mg,0.0469mmol)中。搅拌30min后,将该反应物加热至70℃保持1小时,冷却至室温,并用etoac(10ml)稀释,向其中加入饱和的nahco3,并搅拌30min。将水层分离,并用etoac(1×)萃取;将合并的有机层用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到糊状物,向其中加入nbuoh(5ml)和hoac(0.5ml)。将其在115℃加热16小时,冷却,并在真空中浓缩,用etoac稀释,用饱和nahco3,盐水洗涤,并经mgso4干燥。浓缩的滤液的制备型tlc(5%的meoh在etoac中的溶液)得到11.5mg(51%)作为微黄色固体的期望的三氟甲基噁二唑类似物化合物159。ms:[m+1]=484。
1652、方案25
1653、
1654、实施例89:化合物162的合成:
1655、
1656、在-20℃向在thf(2.9ml)和dmf(0.8ml)中搅拌的内酰胺62(503.4mg,1.42mmol)中加入tbuok(240.2mg)。搅拌30分钟后,逐滴加入氯磷酸二乙酯(377.7mg,2.12mmol),并将该反应混合物在3h内缓慢地温热至8℃,随后冷却至-20℃。加入2.26ml(2.26mmol)噁二唑异氰酸酯(参考jmc,1996,39,170;制备为1m thf溶液)。将该反应混合物进一步冷却至-78℃,加入tbuok(238.4mg),并将该反应物缓慢地温热至室温过夜。加入饱和的nh4cl(5ml),并将该混合物用etoac(2×)萃取,用盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物通过使用0-10%的meoh在etoac中的溶液梯度洗脱的硅胶柱色谱法纯化以得到246.0mg作为微黄色固体的咪唑产物。ms:[m+1]=462。
1657、将以上获得的咪唑(246.0mg,0.533mmol)在dcm(3ml)中搅拌。加入三氟乙酸(3ml),随后加入三氟甲基磺酸(160.0mg,1.07mmol)。搅拌3h后,将该反应物用dcm(20ml)稀释,用饱和的nahco3洗涤;将水层分离,并用dcm(2×)萃取;将合并的dcm溶液用盐水洗涤,并经mgso4干燥。过滤,并在真空中除去溶剂,得到208.7mg作为微黄色絮片状固体的粗制内酰胺产物。[m+1]=312。
1658、在氮气气氛下将磷酰氯(29.9mg,0.195mmol)加入以上得到的内酰胺(22.5mg,0.0723mmol)和n,n-二甲基-对甲苯胺(51.8mg,0.383mmol)在氯苯(0.45ml)中的搅拌溶液中。将该反应混合物在135℃加热3小时,然后冷却至室温。加入二异丙基乙胺(75.7mg,0.586mmol)和苯氧基乙酰肼(50.1mg,0.302mmol),并将该反应混合物在100℃加热14小时,冷却至室温,并在饱和nh4cl和etoac之间分配。将水层分离,并用etoac萃取;将合并的etoac溶液用盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物化合物162通过使用0-10%的meoh在etoac中的溶液梯度洗脱的硅胶柱色谱法分离为微黄色固体。重量:11.8mg(37%)。ms:[m+1]=442。
1659、实施例90:化合物163的合成:
1660、
1661、以与关于实施例89描述的途径类似的合成途径,使用4-氟苯氧基乙酰肼替代苯氧基乙酰肼,合成实施例90的化合物,以得到作为微黄色固体的实施例90的化合物:ms:[m+1]=460。
1662、实施例91:化合物164的合成:
1663、
1664、以与关于实施例89描述的途径类似的合成途径,使用甲氧基乙酰肼替代苯氧基乙酰肼,合成实施例91的化合物,以得到作为微黄色固体的实施例91的化合物:ms:[m+1]=380。
1665、实施例92:化合物165的合成:
1666、
1667、苄氧基乙酰肼的制备:在0℃将羰基二咪唑(1.52g,9.39mmol)加入在thf(60ml)中搅拌的苄氧基乙酸(1.2g,7.22mmol)中。除去冰浴,并继续搅拌1小时。在室温将得到的浑浊的溶液加入在thf(40ml)中搅拌的肼(0.927g,28.9mmol)中。16小时后,将该反应混合物浓缩为浆料,向其中加入水(120ml),用dcm(3×)萃取;将合并的dcm溶液用盐水洗涤,并经mgso4干燥。过滤,并除去溶剂,得到0.908g(70%)作为澄清粘稠油的酰肼。在使用前将其在甲苯中共沸几次。
1668、以与关于实施例89描述的途径类似的合成途径,使用苄氧基乙酰肼替代苯氧基乙酰肼,合成实施例92的化合物,以得到作为微黄色固体的实施例92的化合物:ms:[m+1]=456。
1669、实施例93:化合物166的合成:
1670、
1671、在氢气气氛下将来自以上的化合物165(58.5mg,0.128mmol)用在etoac(4ml)和meoh(4ml)中的10%pd-c(催化)处理2小时。穿过硅藻土过滤催化剂。向滤液中加入浓hcl(0.89ml),并将该混合物在室温搅拌16小时。加入过量的na2co3(水溶液),并将该溶液用etoac(2×)萃取;将合并的有机溶液用盐水洗涤,并经mgso4干燥。使用15%的meoh在etoac中的溶液进行浓缩的滤液的制备型tlc,得到14.9mg作为微黄色固体的伯酰胺([m+1]=417)。在90℃将该伯酰胺用在1,4-二噁烷(1ml)中的磷酰氯(54.9mg,0.358mmol)处理14小时。冷却后,将该反应混合物用etoac稀释,用饱和的nahco3洗涤;将水层分离,并用etoac(1×)萃取,将合并的有机溶液用盐水洗涤,并经mgso4干燥。使用5%的meoh在etoac中的溶液进行浓缩的滤液的制备型tlc,得到5.2mg作为白色针状物的期望的腈产物化合物166。[m+1]=399。
1672、方案26
1673、
1674、实施例94:化合物169的合成:
1675、
1676、在-20℃向在thf(10ml)和dmf(3ml)中搅拌的内酰胺62(2.23g,6.24mmol)中加入tbuok(1.05g,9.36mmol)。搅拌30分钟后,逐滴加入氯磷酸二乙酯(1.66g,9.36mmol),并将该反应混合物在3h内缓慢地温热至8-10℃,随后冷却至-20℃。加入10.0ml(10.0mmol)噁二唑异氰酸酯(参考jmc,1996,39,170;制备为1m thf溶液)。将该反应混合物进一步冷却至-78℃,加入tbuok(1.05g,9.36mmol),并将该反应物缓慢地温热至室温过夜。加入饱和的nh4cl(20ml),并将该混合物用etoac(3×)萃取,用盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物通过使用10-100%的etoac在dcm中的溶液梯度洗脱的硅胶柱色谱法分离,以得到1.07g(35%)作为微黄色泡沫状固体的咪唑产物。ms:[m+1]=490。
1677、将上面得到的咪唑(1.07g,2.18mmol)在dcm(11ml)中搅拌。加入三氟乙酸(11ml),随后加入三氟甲基磺酸(0.656g,4.37mmol)。搅拌4h后,将该反应混合物在真空中浓缩,用dcm(50ml)稀释,用饱和的nahco3洗涤;将水层分离,并用dcm(2×)萃取;将合并的dcm溶液用盐水洗涤,并经mgso4干燥。过滤,并在真空中除去溶剂,得到0.872g作为淡棕色固体的粗制内酰胺产物。[m+1]=340。
1678、在氮气气氛下将磷酰氯(51.0mg,0.333mmol)加入以上得到的内酰胺(45.0mg,0.133mmol)和n,n-二甲基-对甲苯胺(89.6mg,0.663mmol)在氯苯(0.60ml)中的搅拌溶液中。将该反应混合物在135℃加热3小时,然后冷却至室温。加入二异丙基乙胺(137.5mg,1.06mmol)和甲氧基乙酰肼(83.1mg,0.798mmol),并将该反应混合物在100℃加热4小时,冷却至室温,用etoac稀释,用饱和的nahco3,盐水洗涤,并经mgso4干燥。过滤并浓缩后,将产物化合物169通过使用0-13%的meoh在etoac中的溶液梯度洗脱的硅胶柱色谱法分离为淡棕色固体。重量:14.3mg(26%)。ms:[m+1]=408。
1679、实施例95:化合物171的合成:
1680、
1681、以与关于实施例94描述的途径类似的合成途径,使用苯氧基乙酰肼替代甲氧基乙酰肼,合成实施例95的化合物,以得到作为微黄色固体的实施例95的化合物:ms:[m+1]=470。
1682、实施例96:化合物172的合成:
1683、
1684、以与关于实施例94描述的途径类似的合成途径,使用4-氟-苯氧基乙酰肼替代甲氧基乙酰肼,合成实施例96的化合物,以得到作为微黄色固体的实施例96的化合物:ms:[m+1]=488。
1685、实施例97:化合物173的合成:
1686、
1687、以与关于实施例94描述的途径类似的合成途径,使用乙氧基乙酰肼替代甲氧基乙酰肼,合成实施例97的化合物,以得到作为微黄色固体的实施例97的化合物:ms:[m+1]=422。
1688、实施例98:化合物174的合成:
1689、
1690、以与关于实施例94描述的途径类似的合成途径,使用2-氟-苯氧基乙酰肼替代甲氧基乙酰肼,合成实施例98的化合物,以得到作为微黄色固体的实施例98的化合物:ms:[m+1]=488。
1691、实施例99:化合物175的合成:
1692、
1693、以与关于实施例94描述的途径类似的合成途径,使用2-氯-苯氧基乙酰肼替代甲氧基乙酰肼,合成实施例99的化合物,以得到作为微黄色固体的实施例99的化合物:ms:[m+1]=504。
1694、实施例100:化合物176的合成:
1695、
1696、3-吡啶基氧基乙酰肼的制备:将乙基3-吡啶基氧基乙酸酯(0.50g,2.76mmol)和肼(0.31g,9.66mmol)在异丙醇(35ml)中的溶液在85℃加热30小时,冷却,并在真空中浓缩。将得到的白色固体溶解在少量的饱和的nacl溶液中,并用etoac重复萃取。将合并的有机溶液经mgso4干燥。过滤,并除去溶剂,得到177mg作为白色固体的期望的乙酰肼。通过在甲苯中共沸除去残余的水分。
1697、以与关于实施例94描述的途径类似的合成途径,使用3-吡啶基氧基乙酰肼替代甲氧基乙酰肼,合成实施例100的化合物,以得到作为微黄色固体的实施例100的化合物:ms:[m+1]=471。
1698、实施例101:化合物177的合成:
1699、
1700、以与关于实施例94描述的途径类似的合成途径,使用1-萘氧基乙酰肼替代甲氧基乙酰肼,合成实施例101的化合物,以得到作为灰白色固体的实施例101的化合物:ms:[m+1]=520。
1701、实施例102:化合物179的合成:
1702、
1703、以与关于实施例94描述的途径类似的合成途径,使用3-氟苯氧基乙酰肼替代甲氧基乙酰肼,合成实施例102的化合物,以得到作为微黄色固体的实施例102的化合物:ms:[m+1]=488。
1704、实施例103:化合物178的合成:
1705、
1706、在氮气气氛下将磷酰氯(64.8mg,0.422mmol)加入噁二唑基咪唑内酰胺(57.5mg,0.169mmol)和n,n-二甲基-对甲苯胺(114.6mg,0.847mmol)在氯苯(0.70ml)中的搅拌溶液中。将该反应混合物在135℃加热3小时,然后冷却至室温。加入二异丙基乙胺(174.7mg,1.35mmol),叔-buoh(0.3ml)和2-羟基乙酰肼(91.3mg,1.01mmol)。将该反应混合物在室温搅拌20min,然后在50℃温热1小时,随后在80℃加热1小时,最后在100℃加热过夜。冷却至室温后,将该反应物用etoac稀释,用盐水洗涤,并经mgso4干燥。使用0-20%的meoh在etoac中的溶液梯度洗脱进行浓缩的滤液的硅胶柱色谱法,得到作为微黄色固体的期望的羟基甲基三唑产物。重量:18.1mg(27%)。ms:[m+1]=394。
1707、向来自上面的羟基甲基三唑(18.1mg,0.046mmol),环戊基溴(274.0mg,1.84mmol)和hmpa(16.5mg,0.092mmol)在thf(0.5ml)中的搅拌溶液中加入nah(60%悬浮液;18.4mg,0.46mmol)。10min后,将该反应物在100℃加热6小时,冷却,用饱和的nahco3猝灭,用etoac(2×)萃取,用盐水洗涤,并经mgso4干燥。使用8%的meoh在etoac中的溶液进行浓缩的滤液的制备型tlc,得到5.5mg(26%)作为微黄色固体的期望的醚化合物178。[m+1]=462。
1708、方案27
1709、
1710、实施例104:化合物168的合成:
1711、
1712、向甘氨酸苄酯盐酸盐(5g,24.8mmol)在dcm(100ml)中的悬浮液中加入edc.hcl(6.2g,33.2mmol)和三乙胺(5.2ml,37.2mmol)。将该悬浮液冷却至-50℃,然后加入甲酸(1.4ml,37.2mmol)在dcm(5ml)中的溶液。将该反应混合物在-50℃搅拌1小时,然后在4℃搅拌3小时。将该溶液用1n hcl稀释,并用dcm(2×)萃取。将合并的有机相用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到3.89g(81%收率)作为油的甲酰基化的甘氨酸(m+1=194)。
1713、向甲酰基化的甘氨酸衍生物(1g,5.2mmol)在dcm(30ml)中的溶液中加入三乙胺(3.2ml,23mmol)。将该溶液冷却至-50℃,并缓慢地加入pocl3(1.9ml,20.8mmol)。将该溶液在-50℃搅拌10分钟,然后在室温搅拌40分钟。该溶液变为淡棕红色。将其用dcm稀释,并加入20%碳酸钠溶液(100ml)。将该反应混合物剧烈搅拌15分钟。将有机相分离两次,并经mgso4干燥。过滤并浓缩,以定量收率得到期望的异氰基乙酸苄酯,将其不经进一步纯化地用于下一步。
1714、在0℃向1,2,4-三唑(914mg,13.2mmol)在无水ch3cn(20ml)中的溶液中加入i-pr2net(2.5ml,14.6mmol)。一旦所有三唑被溶解,加入pocl3(0.43ml,4.6mmol)。将该混合物在0℃搅拌2小时。加入内酰胺酯16’(1g,3.31mmol)。将得到的溶液在油浴中在80℃加热16小时。将该混合物用冰浴冷却。用etoac稀释,然后加入水。将水层分离,并用etoac萃取四次。将合并的有机萃取物用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到浅黄色固体,将其直接用在下一步中(m+1=354)。
1715、在氮气气氛下将异氰基乙酸苄酯(892mg,5.1mmol)在dmf(10ml)中的溶液冷却至-50℃。加入kotbu(514mg,4.6mmol)。将该混合物在-50℃搅拌1小时。在-50℃缓慢地加入在dmf(5ml)中的以上制备的三唑衍生物(900mg,2.55mmol)。历时16h使该混合物温热至室温。加入饱和的nh4cl水溶液,并将其用etoac萃取三次。将合并的萃取物用盐水(3×)洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。色谱法(redisep24g硅胶柱,用70%的etoac在己烷类中的溶液洗脱)得到886mg(76%收率)的产物(m+1=460)。
1716、向苄酯衍生物(770mg,1.68mmol)在etoac(10ml)和meoh(30ml)中的溶液中加入湿的pd/c(60mg),并将得到的悬浮液在氢气气氛下搅拌48小时。将该悬浮液穿过硅藻土过滤,并将得到的溶液浓缩。将粗制的去苄基化的产物(530mg,86%收率)不经进一步纯化地用于下一步(m+1=370)。
1717、向酸(530mg,1.44mmol)在dcm(10ml)中的悬浮液中加入cdi(931mg,5.75mmol)。将该溶液在室温搅拌2小时。将该溶液用冰浴冷却,并加入nh4oh溶液(6ml)。将该溶液搅拌30分钟,并将其浓缩。将固体通过过滤收集,并用水洗涤,得到422mg(80%)作为棕色固体的期望产物。(m+1=369)。
1718、向伯酰胺衍生物(422mg,1.15mmol)在二噁烷(10ml)中的悬浮液中加入pocl3(160μl,1.7mmol)。将该悬浮液在90℃加热2小时。将得到的溶液用冰浴冷却,并用饱和的nahco3水溶液猝灭。将固体通过过滤收集,得到308mg(77%收率)的期望的氰化物衍生物。(m+1=351)。
1719、向氰化物衍生物(150mg,0.44mmol)在etoh(4ml)和水(1ml)中的悬浮液中加入盐酸羟胺(40mg,0.57mmol)和碳酸钾(67mg,0.48mmol)。将该悬浮液在室温搅拌16小时。lcms指示约50%转化率。加入更多的盐酸羟胺(40mg,0.57mmol)和碳酸钾(67mg,0.48mmol),并搅拌另外24小时。将该溶液用etoac稀释,并用水洗涤。将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到145mg(86%收率)的期望产物。(m+1=384)。
1720、向乙酸(0.22ml,3.8mmol)在thf(5ml)中的溶液中加入cdi(123mg,0.76mmol)。将该溶液在室温搅拌2小时。然后将该溶液倒入含肟衍生物(145mg,0.38mmol)的烧瓶中,并在70c加热1小时。将溶剂蒸发,并将粗制物质悬浮于乙酸(8ml)中,并在130℃加热1小时。将溶剂蒸发,并将粗制物质与水一起研磨,得到134mg(86%)期望产物(m+1=408)。
1721、向酯衍生物(50mg,0.12mmol)在thf(1ml)中的悬浮液中加入氢化铝锂(7mg,0.18mmol)。将该悬浮液在室温搅拌2小时。lcms指示约70%转化率,伴有一些其它的副产物和一些剩余的起始原料。加入更多氢化铝锂(4mg),并将该反应混合物在室温搅拌另外30分钟。将该反应混合物用1n hcl猝灭。将该溶液用etoac(3×)萃取。将合并的有机相用盐水洗涤,经mgso4干燥。过滤并浓缩,得到20mg(45%收率)的期望的醇产物。(m+1=366)。
1722、向醇(20mg,0.055mmol)在二噁烷(1ml)中的悬浮液中加入pobr3(31mg,0.11mmol)。将该反应混合物在110℃加热1小时。将该反应混合物用冰浴冷却,并加入饱和的nahco3水溶液。将得到的溶液用etoac(3×)萃取。将合并的有机相用盐水洗涤,并经mgso4干燥。将溶剂浓缩,得到22mg(96%收率)的期望产物(m+1=428)。
1723、向含烷基溴衍生物(22mg,0.052mmol)的小瓶中加入3-氟苯酚(58mg,0.52mmol)在二噁烷(1ml)中的溶液和碳酸钾(72mg,0.52mmol)。将该反应混合物在90℃加热1小时。将该反应混合物用饱和的nahco3水溶液稀释。将得到的溶液用etoac(3x)萃取。将合并的有机相用盐水洗涤,并经mgso4干燥。过滤并浓缩,得到粗产物。通过制备型tlc(洗脱系统:etoac)纯化以得到5mg(21%收率)的期望产物化合物168(m+1=460)。h1nmr(cdcl3)δ7.87(1h,s),7.65(1h,d,j=3.5hz),7.57(1h,d,j=10hz),7.24(1h,m),7.19(1h,dd,j=3.5,9hz),6.77(1h,dd,j=2.5,9.5hz),6.72(2h,m),5.26(2h,s),3.97(3h,s),2.48(3h,s)。
1724、化合物215-313的合成
1725、方案28
1726、
1727、中间体a(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸乙酯)的合成。
1728、在氮气气氛下在0℃将溴乙酸乙酯(方案28)(10.0克,59.87mmol)在20.0ml无水thf中的溶液逐滴加入(2,4-二甲氧基苄基)胺(10.0克,59.81mmol)和三乙胺(6.06克,59.87mmol)在无水thf(20.0ml)中的溶液中。将反应混合物温热至室温并搅拌过夜。加入~100ml盐水,并将反应混合物用乙酸乙酯(2×~100ml)萃取。将合并的萃取物经无水mgso4干燥并在减压下浓缩。使用combiflash色谱法(梯度:20:80至50:50v/v乙酸乙酯:己烷)进行纯化。得到7.6克(收率50.2%)作为无色液体的烷基化产物。c13h19no4[m+h]+的m/z计算值:254;得到:254.1。
1729、将该酯(7.5克,29.6mmol)溶解在40.0ml甲醇中。将反应混合物冷却,并逐滴加入2n naoh(88.82mmol,44.0ml)水溶液。将反应混合物温热至室温并搅拌2小时。将该反应混合物用~75.0ml水稀释,在冰浴中冷却,使用2n的hcl水溶液中和至~5.0-4.5的ph。将多余的水在减压下浓缩,并风干以得到白色固体粉末。将固体溶解在85:15v/v的dcm:meoh(100.0ml)中并过滤,将滤液蒸发以得到7.1克作为白色粉末(吸湿)的羧酸。c11h15no4[m+na]+的m/z计算值:248;得到:248.1。
1730、将以上化合物(7.0克,31.08mmol)和6.14克,31.08mmol的5-氯靛红酸酐在70.0ml对二甲苯中混合,并在140℃温度回流3小时。将反应混合物过滤并将粗产物从甲醇中重结晶。得到8.5克作为白色粉末的7-氯-4-[(2,4-二甲氧基苯基)甲基]-2,3,4,5-四氢-1h-1,4-苯并二氮杂环庚三烯-2,5-二酮(75.8%收率)。c18h17cln2o4[m+h]+的m/z计算值:361;得到:361.1。
1731、将以上苯并二氮杂环庚三烯-2,5-二酮(4克,11.1mmol)溶解在thf/dmf(57.2/12.7ml)中并在-20℃温度冷却。加入精细粉碎的叔丁醇钾粉末(1.9克,16.6mmol),并将反应混合物在-20℃搅拌20.0分钟。在-20℃将3.1克,17.7mmol的氯代磷酸二乙酯逐滴加入反应混合物中,并达到0-5℃保持3小时。将反应混合物在环境温度搅拌10.0分钟。在-20℃将2.1克,18.4mmol的异氰基乙酸乙酯加入反应混合物中,并将反应混合物进一步冷却至-78℃。在-78℃加入1.9克,16.6mmol的精细粉碎的叔丁醇钾粉末,并将该反应混合物通过缓慢地温热至环境温度搅拌过夜。将反应混合物用饱和nh4cl水溶液(10ml)猝灭,用乙酸乙酯(3×20ml)萃取。将合并的萃取物经无水mgso4干燥并在减压下浓缩。将粗产物从乙酸乙酯中重结晶以得到2.2克的作为白色固体的12-氯-8-[(2,4-二甲氧基苯基)甲基]-9-氧代-2,4,8-三氮杂三环[8.4.0.02,6]十四烷-1(14),3,5,10,12-戊烯-5-甲酸乙酯。从母液得到第二批,以得到另外3.5g产物(64%收率)。
1732、通过将以上化合物(2.2克,4.83mmol)溶解在dcm(25.0ml)中除去二甲氧基苄基保护基,随后加入25.0ml三氟乙酸和1.45克,9.65mmol的三氟甲磺酸。将反应混合物在室温搅拌90分钟。将反应混合物用nahco3水溶液中和,并将沉淀物过滤,用水洗涤并干燥以得到1.9克作为固体产物的12-氯-9-氧代-2,4,8-三氮杂三环[8.4.0.02,6]十四烷-1(14),3,5,10,12-戊烯-5-甲酸乙酯。c14h12cln3o3[m+h]+的m/z计算值:306;得到:306.1。
1733、在第一步中,将来自上面的12-氯-9-氧代-2,4,8-三氮杂三环[8.4.0.02,6]十四烷-1(14),3,5,10,12-戊烯-5-甲酸乙酯(1.9克,6.21mmol)溶解在25.0ml氯苯中,随后加入2.52克,18.64mmol的4,n,n-三甲基苯胺,1.42克,9.32mmol的pocl3,并将反应混合物在135℃回流2小时。lcms指示~50%起始原料保持未反应。在室温将1.68克,12.42mmol的另外的4,n,n-三甲基苯胺和0.95克,6.21mmol的pocl3进一步加入反应混合物中,并在135℃回流1小时。lcms指示~10%起始原料保持未反应。在室温将另外0.84克,6.21mmol的4,n,n-三甲基苯胺(总共6.0当量)和0.48克,3.11mmol的pocl3(总共3当量)进一步加入反应混合物中,并在135℃回流1小时。
1734、在第二步中,在室温将4.67克,44.75mmol的甲氧基乙酸酰肼(总共7.2当量),随后7.71克,59.66mmol的n,n-二异丙基乙胺加入反应混合物中,并在100℃回流1小时。将反应混合物冷却至室温并用nahco3水溶液(~25.0ml)中和。将有机物用乙酸乙酯(75ml×3),随后用dcm(50.0ml×3)萃取,并用盐水洗涤。将etoac有机层通过过滤不溶性沉淀物(0.805克纯产物)分离,并将合并的有机层经无水mgso4干燥,在减压下浓缩。将粗产物通过combiflash色谱法(流动相:0-10%meoh:etoac)纯化以产生另外0.8克黄色固体。中间体a(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]-十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸乙酯)的最后两步的总收率是72.58%。c17h16cln5o3[m+h]+的m/z计算值:374;得到:374.1。
1735、中间体b(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸)的合成。
1736、将中间体a(0.4克,1.07mmol)溶解在thf/h2o/meoh(3.2/4.8/8.0v/v ml)的混合物中。将0.05克,2.14mmol的lioh加入,并将反应混合物在室温搅拌3小时。将反应混合物用2n hcl水溶液酸化,将沉淀物收集,并用去离子水洗涤。干燥以后,得到0.36克作为白色固体的中间体b(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸)。c15h12cln5o3[m+h]+的m/z计算值:346;得到:345.9。
1737、中间体c(15-氯-9-(苯氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸乙酯)和d(15-氯-9-(苄氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸乙酯)与中间体a类似地合成,分别使用2-苯氧基乙酰肼和2-(苄氧基)乙酰肼代替2-甲氧基乙酰肼。
1738、方案29解释了使用中间体a以产生新类似物的一些选择的实施例。
1739、方案29
1740、
1741、化合物233的合成:
1742、
1743、将丙酮肟(0.22克,0.31mmol)溶解在无水thf(0.5ml)中。逐滴加入0.38ml,0.62mmol的1.6m正-buli,并将反应混合物在单独烧瓶中在0-5℃搅拌1小时。在0-5℃通过插管加入中间体a(0.05克,0.13mmol)在1.0ml thf中的溶液,并将反应物通过在室温逐渐温热而搅拌16小时。lcms指示起始原料和中间体m/z:374.1(~45/14%,两个峰合并),m/z401(~10%),m/z 402(18%)。
1744、将反应混合物用0.03ml浓h2so4,随后用0.03ml去离子水猝灭,并回流2小时。lcms指示起始原料,产物和中间体m/z:374.1(~43%),m/z 383(~40%),m/z 402(17%)。
1745、将反应混合物在减压下浓缩并用nahco3水溶液中和,将沉淀物收集,并用去离子水洗涤。干燥以后,得到22.0mg粗制沉淀物。将化合物使用1:99meoh:chcl3通过制备型tlc板纯化。
1746、化合物238的合成:
1747、
1748、步骤1:将中间体a(0.045克,0.12mmol)溶解在无水甲苯(3.0ml)中。将0.05ml,0.25mmol的氨基乙醇(35.0当量)加入,并将反应混合物回流16小时。将甲苯蒸发,并将反应混合物溶解在dcm(25.0ml)中。将dcm层用盐水,随后用去离子水洗涤,分离,并经无水mgso4干燥。蒸发有机层,得到38.3mg对应的酰胺。lcms指示产物形成m/z:389。
1749、步骤2:将以上酰胺(0.038克,0.09mmol)溶解在无水dcm(2.0ml)中。在0℃温度将0.026ml,0.2mmol的dast(2.0当量)加入反应混合物中并在0℃搅拌1.5小时。在0℃将0.065克固体k2co3(4.8当量)加入,并将反应混合物搅拌30分钟。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发溶剂得到36mg白色固体产物。c17h15cln6o2[m+h]+的m/z计算值:371;得到:371。
1750、化合物239的合成:
1751、
1752、步骤1:将中间体a(0.05克,0.13mmol)溶解在无水甲苯(3.0ml)中。将0.28克,2.67mmol的氨基乙醇(20.0当量)加入,并将反应混合物回流16小时。将甲苯蒸发,并将反应混合物溶解在dcm(25.0ml)中。将dcm层用盐水,随后用去离子水洗涤,分离并经无水mgso4干燥。蒸发有机层,得到酰胺。lcms指示产物形成m/z:431
1753、步骤2:将以上酰胺(0.057克,0.13mmol)溶解在无水dcm(2.0ml)中。在0℃温度将0.035ml,0.3mmol的dast(2.0当量)加入反应混合物中并在0℃搅拌1.5小时。lcms指示产物形成m/z 413。在0℃将0.088克固体k2co3(4.8当量)加入,并将反应混合物搅拌30分钟。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。浓缩有机层,得到产物,将其与20/80hex/etoac一起研磨以得到固体,将其通过过滤收集并干燥:49.4mg(89%)。
1754、化合物243的合成:
1755、
1756、步骤1:将中间体a(0.05克,0.13mmol)溶解在无水甲苯(3.0ml)中。将0.02ml,2.67mmol的氨基醇(20.0当量)加入,并将反应混合物回流16小时。lcms指示起始原料剩余。放入二甲苯(3.0ml)并加入10.0当量的3-氨基丁-1-醇,并将反应混合物回流16小时。最后需要总共40.0当量的氨基乙醇以将所有起始原料转化成回流的二甲苯中的产物。将反应混合物冷却至0℃并将沉淀物过滤。将滤液用dcm(15.0ml×4)萃取。将dcm层用盐水,随后用去离子水洗涤,分离并经无水mgso4干燥。蒸发有机层,得到对应的酰胺。lcms指示产物形成m/z:403。
1757、步骤2:将以上酰胺(0.054克,0.13mmol)溶解在无水dcm(2.0ml)中。在0℃温度将0.05ml,0.33mmol的dast加入反应混合物中,并在0℃搅拌1.5小时。lcms指示产物形成。在0℃加入0.09克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发溶剂得到粗产物。通过制备型tlc(流动相:95:05,dcm:meoh)进行纯化。c18h17cln6o2[m+h]+的m/z计算值:385;得到:385。
1758、化合物244的合成:
1759、
1760、将来自上面的化合物243(0.011克,0.03mmol)溶解在甲苯(2.0ml)中。加入0.010克,0.04mmol的ddq,并将反应混合物在50℃搅拌1小时。lcms指示起始原料m/z 385和小量的产物m/z 383。将反应混合物在60℃搅拌3小时。lcms指示起始原料m/z 385,产物m/z383。将反应混合物在70℃搅拌2小时。lcms指示起始原料m/z 385,产物m/z383和副产物m/z421。将反应混合物在40℃搅拌16小时。lcms指示大量的产物m/z 383和小量的副产物m/z421和起始原料。将甲苯蒸发,并将粗产物通过制备型tlc板(流动相dcm:meoh,95:05v/v)纯化以得到4.4mg产物。c18h15cln6o2[m+h]+的m/z计算值:383;得到:383。
1761、化合物249的合成:
1762、
1763、将来自上面的化合物238(0.016克,0.05mmol)溶解在甲苯(2.0ml)中。加入0.015克,0.07mmol的ddq,并将反应混合物在50℃搅拌1小时。lcms指示起始原料m/z 371。将反应混合物在60℃搅拌5小时。lcms指示起始原料m/z 371,产物m/z 369和不希望的物质m/z407。将反应混合物在30℃搅拌16小时。lcms指示起始原料m/z 371,产物m/z 369和副产物m/z 407。将反应混合物在65℃搅拌4小时。lcms指示产物m/z 369,副产物m/z 407和小量的起始原料。将甲苯蒸发,并将粗产物通过制备型tlc板(流动相dcm:meoh,95:05v/v)纯化以得到2.3mg产物。c17h13cln6o2[m+h]+的m/z计算值:369;得到:369。
1764、化合物256的合成:
1765、
1766、步骤1:将中间体a(0.1克,0.27mmol)溶解在无水thf(3.0ml)中。逐滴加入0.67ml,0.67mmol的1.0m的dibal在thf中的溶液,并将反应混合物在0-5℃搅拌2小时。lcms指示醇还原产物形成m/z332。将反应物用meoh(1.0ml),然后用水(0.5ml)猝灭。加入nahco3的饱和溶液,并将沉淀物穿过硅藻土床过滤。将产物使用dcm(25.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。蒸发溶剂得到46.1mg作为固体产物的[15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-基]甲醇,收率51.9%。c15h14cln5o2[m+h]+的m/z计算值:332;得到:332。
1767、步骤2:将以上醇(0.05克,0.14mmol)溶解在无水dcm(3.0ml)中。加入0.09克的戴斯-马丁过碘烷,并将反应混合物在室温搅拌2小时。lcms指示产物形成m/z 330。将反应物用1n naoh溶液(2-ml)猝灭。加入饱和nahco3溶液,并将产物使用dcm(20.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。蒸发溶剂得到作为固体产物的期望的醛(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛),收率定量。c15h12cln5o2[m+h]+的m/z计算值:330;得到:330。
1768、步骤3:在-78℃温度将1.6m正-buli在己烷中的溶液(0.68ml,1.08mmol)逐滴加入1.4ml,0.86mmol的三甲基甲硅烷基重氮甲烷在己烷中的溶液(溶解在3.0ml thf中)。将反应混合物在-78℃温度搅拌30.0分钟。在-78℃温度将在步骤2中得到的醛(0.142克,0.43mmol)在3.0ml thf中的溶液逐滴加入反应混合物中并逐渐温热至室温。lcms指示产物形成m/z 326和起始原料m/z 330。将反应混合物用饱和的nh4cl溶液猝灭。将产物使用dcm(15.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。通过iscocombiflash纯化系统(流动相:乙酸乙酯/己烷)执行粗产物的纯化。得到19.0mg化合物256,并分离出71.6mg起始原料。c16h12cln5o[m+h]+的m/z计算值:326;得到:326。
1769、化合物285的合成:
1770、
1771、将化合物256(0.025克,0.08mmol)溶解在脱气的dmf(2.0ml)中。将0.03ml,0.23mmol的碘苯加入反应混合物中,随后加入0.06ml,0.41mmol的tea。将反应混合物在室温搅拌。将0.04克,0.04mmol的pd(pph3)4和0.003克,0.015mmol的cui混合物加入反应混合物中并搅拌16小时。lcms指示产物形成m/z 402。将该反应混合物用去离子水稀释。将产物使用dcm(10.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。将粗制反应混合物通过制备型tlc板(流动相:etoac/meoh)纯化。c22h16cln5o[m+h]+的m/z计算值:402;得到:402。
1772、化合物314的合成:
1773、
1774、将来自化合物256的合成步骤2的醛(15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛)(0.015g,0.04mmol)和0.011g(0.055mmol)的tosmic溶于meoh(2.5ml)。加入0.013g(0.09mmol)的k2co3和将反应混合物在60℃搅拌2小时。lcms显示产品形成m/z 369.1。蒸发meoh,将ppts溶于水,用2n hcl水溶液酸化。过滤ppts,用去离子水洗涤,提供10.4mg(62%)的化合物314;c17h13cln6o2[m+h]+的m/z计算值:369;得到:369.1。
1775、化合物339的合成:
1776、
1777、co化m合po物u nd 339
1778、在氮下将1,3-二(1-金刚烷基)咪唑鎓氯化物(0.07g,0.01mmol),[(烯丙基)pdcl]2(0.003g,0.009mmol),cui(0.004g,0.02mmol)和cs2co3(0.04g,0.13mmol)加入小瓶。加入二乙醚和dmf(2:1,2.0ml)的混合物,随后是0.03gm(0.09mmol)化合物256和0.014g(0.1mmol)mei。在40.0℃温度搅拌反应混合物16小时。lcms显示产品形成m/z340.2。反应混合物用水(20.0ml)淬灭,用乙酸乙酯(40.0ml)稀释。过滤经合并的层。分开有机层,在无水na2so4上干燥。蒸发有机层,获得粗制产品。粗制产品的纯化通过制备型tlc板进行:流动相:etoac:meoh,97:03v/v ml,提供8.5mg化合物339(27%):
1779、c17h14cln5o[m+h]+的m/z计算值340,得到340.2。
1780、化合物345的合成:
1781、
1782、化合物345以与化合物339(方案29)类似的方式制备,用碘乙烷代替碘甲烷,提供化合物,c18h16cln5o[m+h]+的m/z计算值354,得到354.2。
1783、化合物346的合成:
1784、
1785、化合物346以与化合物339(方案29)类似的方式制备,用苄基溴代替碘甲烷,提供化合物,c23h18cln5o[m+h]+的m/z计算值416,得到416.3
1786、化合物329的合成:
1787、
1788、醇[15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环-[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-基]甲醇
1789、化合物254的合成:
1790、
1791、将异丁腈(10.0克,144.70mmol)溶解在etoh:水(150:50ml,v/v)中,随后加入10.0克,144.70mmol的盐酸羟胺和20.0克,144.70mmol的k2co3。将反应混合物在80℃回流6小时。将溶剂在减压下蒸发,并将得到的固体用150ml乙醇处理,声处理,过滤并用100ml乙醇洗涤。将合并的滤液在减压下蒸发,并与甲苯(25.0ml×3)一起共沸以得到8.1克作为无色液体浆料的n'-羟基-2-甲基丙脒(54.8%收率)。将以上酰胺-肟(1.37克,13.38mmol)在使用前与甲苯(10ml×5)一起共沸,并溶解于20.0ml无水thf中。在0℃将0.27克,6.69mmol的nah分成三份加入反应混合物中,并在环境温度搅拌30.0分钟。将0.5克,1.34mmol的中间体a加入,并将反应混合物在环境温度搅拌45.0分钟,并在67℃回流90.0分钟。将溶剂在减压下蒸发,并将得到的黄色糊状物用25.0ml饱和nahco3水溶液处理。将沉淀物穿过漏斗过滤,并用10.0ml水和10.0ml己烷洗涤以得到0.380克固体(69.1%收率)。c19h18cln7o2[m+h]+的m/z计算值:412.0;得到:412.1。
1792、化合物215的合成
1793、
1794、将醇[15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-基]甲醇(在化合物256,步骤1中制备)(34mg,0.1025mmol)悬浮于无水thf(2ml)中。加入hmpa(36.7mg,0.205mmol),随后加入乙基碘(0.33ml)和nah(41mg 60%的在油中的悬浮液)。将该反应物在室温搅拌5分钟,然后加热至70℃过夜。将混合物冷却,并在etoac和盐水之间分配。将有机相干燥并浓缩以得到油,将其通过柱色谱法(0%至10%的meoh在dcm中的溶液)纯化以得到3.7mg作为油的化合物215。
1795、化合物274的合成
1796、
1797、将[15-氯-9-(甲氧基甲基)-2,4,8,10,11五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-基]甲醇(0.02克,0.06mmol)溶解在无水thf(3.0ml)中。加入0.003克的nah,并将反应混合物在室温搅拌30.0分钟。逐滴加入0.012ml,0.12mmol的2-溴吡啶,并将反应混合物在室温搅拌16小时。将反应混合物回流另外2小时。lcms指示m/z409。将该反应物在减压下浓缩并用饱和nahco3溶液稀释。将产物使用dcm(10.0ml×4)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。得到~1.0mg产物。c20h17cln6o2[m+h]+的m/z计算值:409;得到:409。
1798、方案30解释了使用中间体b来制备新类似物的一些选择的实施例。
1799、方案30
1800、草酰氯
1801、
1802、化合物234的合成:
1803、
1804、将中间体b(0.043克,0.12mmol),0.3mmol的edc.hcl和0.048克,0.31mmol的hobt水合物溶解在thf/dcm(1:1,v/v 1.5ml)中,随后加入0.09ml,0.62mmol的三甲基苯胺和0.016ml,0.25mmol的炔丙胺。将反应混合物在室温搅拌16小时。将该反应混合物用氯化铵水溶液稀释,并用乙酸乙酯萃取。将合并的层用盐水洗涤,分离并经无水mgso4干燥。蒸发有机层,得到~13.0mg粗产物。将粗产物通过制备型tlc板(流动相:5:95,meoh,乙酸乙酯)纯化。c18h15cln6o3[m+h]+的m/z计算值:383;得到:383.1
1805、化合物348的合成:
1806、步骤1:以与化合物234的合成类似的方式,将中间体b转化为15-氯-n-(2-羟基苯基)-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酰胺。
1807、步骤2:将上述酰胺(0.017g,0.038mmol)溶于无水甲苯(2.5ml)。将对-甲苯磺酸一水合物(0.043g,0.23mmol)加入,将反应混合物回流16小时。lcms显示~50:50比率的产品和原料。将甲苯替换为二甲苯(2.5ml),反应混合物在130℃加热6小时。lcms显示产品形成m/z 419.2。浓缩反应混合物,用乙酸乙酯稀释(25.0ml)。有机层用nahco3饱和溶液随后盐水洗涤。分开有机层,在无水na2so4上干燥。蒸发溶剂,提供粗制产品。粗制产品的纯化通过制备型tlc板进行,流动相:etoac:meoh,95:05v/v,提供化合物348。
1808、化合物240的合成:
1809、
1810、将步骤1中间体b(0.05克,0.15mmol)溶解在无水dcm(2.0ml)中。加入0.05ml,0.36mmol的三甲胺(2.5当量),随后加入0.024ml,0.29mmol的草酰氯(2.0当量),并将反应混合物在室温搅拌60分钟。在0℃将0.076ml,0.7mmol的氨基醇(5.0当量)加入反应混合物中并搅拌2.5小时。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将合并的有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发有机层,得到54.1mg酰胺。lcms指示产物形成m/z:431
1811、步骤2将(2s)-2-氨基-3-甲基丁基15-氯-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酸酯(0.027克,0.06mmol)溶解在无水dcm(2.0ml)中。在0℃温度将0.016ml,0.13mmol的dast加入反应混合物中,并在0-5℃搅拌3小时。lcms指示产物形成。在0℃加入0.04克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发溶剂得到粗产物。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。得到23.7mg固体产物。质量。c20h21cln6o2[m+h]+的m/z计算值:413;得到:413。
1812、化合物246的合成:
1813、
1814、以与化合物245类似的方式,在50c使用ddq,甲苯将化合物240转化成化合物246,以得到5.5mg(37%)化合物246。lcms指示产物形成m/z:411。
1815、化合物242的合成:
1816、
1817、步骤1:将中间体b(0.025克,0.07mmol)溶解在无水dcm(2.0ml)中。加入0.03ml,0.21mmol的三甲胺(3.0当量),随后加入0.015ml,0.18mmol的草酰氯(2.5当量),并将反应混合物在室温搅拌60分钟。在0℃将0.05克,0.36mmol的(r,s)-2-氨基-2-苯基乙烷-1-醇(5.0当量)加入反应混合物中并在室温搅拌2.5小时。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将合并的有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发有机层,得到期望的酰胺。lcms指示产物形成m/z:465
1818、步骤2:将以上酰胺(0.034克,0.07mmol)溶解在无水dcm(2.0ml)中。在0℃温度将0.03ml,0.22mmol的dast加入反应混合物中并在0℃搅拌1.5小时。lcms指示产物形成。在0℃加入0.05克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发溶剂得到粗产物。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。c23h19cln6o2[m+h]+的m/z计算值:447;得到:447。
1819、化合物245的合成:
1820、
1821、将化合物242(0.015克,0.03mmol)溶解在甲苯(1.5ml)中。加入0.009克,0.04mmol的ddq,并将反应混合物在50℃搅拌1.5小时。lcms指示1:3比率的起始原料m/z 447和产物m/z 445。进一步加入0.005克,0.022mmol的ddq,并将反应混合物在50℃搅拌1.5小时。1:6比率的起始原料m/z 447和产物m/z 445。将反应混合物在室温搅拌16小时。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。将具有m/z:445的带分离,并得到9.3mg固体化合物(收率62.4%)。c23h17cln6o2[m+h]+的m/z计算值:445;得到:445。
1822、化合物237的合成:
1823、
1824、步骤1:将中间体b(0.025克,0.07mmol)溶解在无水dcm中。加入0.009ml,0.02ml,0.14mmol的三甲胺,随后加入0.11mmol的草酰氯,并将反应混合物在室温搅拌30分钟。在0℃将0.028ml,0.36mmol的3-氨基-1-丙醇加入反应混合物中并搅拌2.5小时,然后浓缩。lcms指示产物形成m/z:403,几乎没有起始原料剩余。
1825、步骤2:将来自步骤1的粗制酰胺(0.018克,0.045mmol)溶解在无水dcm(2.0ml)中。在-78℃温度将0.012ml,0.09mmol的dast加入反应混合物中并逐渐温热至0℃。在-78℃加入0.03克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水mgso4干燥。蒸发溶剂得到14.7mg作为白色固体产物的化合物237。c18h17cln6o2[m+h]+的m/z计算值:385;得到:385.1。
1826、化合物263的合成:
1827、
1828、步骤1:将中间体b(0.03克,0.09mmol),0.034克,0.17mmol的edc.hcl和0.027克,0.17mmol的hobt.xh2o溶解在无水dcm(2.5ml)中。加入0.024克,0.17mmol的r-(-)-2-苯基甘氨醇(phenylglycinol),并将反应混合物在室温搅拌6小时。lcms指示产物形成m/z464.9。将反应混合物用去离子水稀释,并用dcm(10.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。蒸发有机层,得到粗产物。得到液体浆状物。c23h21cln6o3[m+h]+的m/z计算值:465;得到:464.9。
1829、步骤2:将以上酰胺(0.04克,0.086mmol)溶解在无水dcm(2.0ml)中。加入0.03ml,0.21mmol的dast,并将反应混合物在0℃温度搅拌2小时。lcms指示产物形成m/z 446.9。在0℃加入0.06克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水na2so4干燥。蒸发溶剂得到粗产物。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。得到25.0mg固体产物。c23h19cln6o2[m+h]+的m/z计算值:447;得到:446.9。
1830、化合物264的合成:
1831、
1832、步骤1:将中间体b(0.03克,0.09mmol),0.034克,0.17mmol的edc.hcl和0.027克,0.17mmol的hobt.xh2o溶解在无水dcm(2.5ml)中。加入0.024克,0.17mmol的s-(+)-2-苯基甘氨醇,并将反应混合物在室温搅拌6小时。lcms指示产物形成m/z 464.9。将反应混合物用去离子水稀释,并用dcm(10.0ml×3)萃取。将合并的dcm层用盐水洗涤,分离并经无水na2so4干燥。蒸发有机层,得到粗产物。得到液体浆状物。c23h21cln6o3[m+h]+的m/z计算值:465;得到:464.9。
1833、步骤2:将以上酰胺(0.04克,0.086mmol)溶解在无水dcm(2.0ml)中。加入0.03ml,0.21mmol的dast,并将反应混合物在0℃温度搅拌2小时。lcms指示产物形成m/z 446.9。在0℃加入0.06克固体k2co3,并将反应混合物逐渐温热至室温。将该反应混合物用nahco3水溶液稀释,并用dcm(15.0ml×3)萃取。将有机层用盐水洗涤,分离并经无水na2so4干燥。蒸发溶剂得到粗产物。通过制备型tlc(流动相:95:05,dcm:meoh)执行纯化。得到26.4mg固体产物。c23h19cln6o2[m+h]+的m/z计算值:447;得到:446.9。
1834、化合物332,334,335,336,337和338用与方案30中描述用于合成化合物264的合成程序类似的合成程序制备。
1835、使用与在方案27中描述的用于合成化合物168的程序类似的合成程序,制备化合物180,181和182。
1836、使用与在方案26中描述的用于合成化合物169-179的程序类似的合成程序,制备化合物183-193。
1837、使用与在方案21和22中描述的程序类似的合成程序,制备化合物194和195。
1838、使用与在方案18a中描述的程序类似的合成程序,制备化合物196-198和206。
1839、使用与在方案18a中描述的用于合成化合物129的程序类似的合成程序,制备化合物202。
1840、使用与在方案18b中描述的程序类似的合成程序,制备化合物199,200,204和205。
1841、使用与在方案24中描述的程序类似的合成程序,制备化合物201和203。
1842、使用与在方案17中描述的程序类似的合成程序,制备化合物207-210。
1843、与在方案22中所示的那些转化类似地,产生化合物207-210中的腈取代基。
1844、使用与在方案20中描述的程序类似的合成程序,制备化合物211-214。
1845、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物255;类似于化合物254。
1846、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物259;类似于化合物243。
1847、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物260;类似于化合物242。
1848、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物261;类似于化合物256。
1849、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物265;类似于化合物264。
1850、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物266;类似于化合物264。
1851、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物267;类似于化合物264。
1852、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物268;类似于化合物263。
1853、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物270;类似于化合物264。
1854、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物271;类似于化合物264。
1855、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物275;类似于化合物264。
1856、使用在方案28和30中描述的合成途径从适当的起始原料制备化合物276;类似于化合物245。
1857、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物278;类似于化合物233。
1858、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物281;类似于化合物233。
1859、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物282,283,286,287;类似于化合物243。
1860、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物288;类似于化合物256。
1861、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物293;类似于化合物285。
1862、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物294,295和296;类似于化合物243和244。
1863、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物303;类似于化合物233。
1864、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物304;类似于化合物264。
1865、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物297;类似于化合物243。
1866、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物307;类似于化合物285。
1867、使用在方案28中描述的合成途径从适当的起始原料制备化合物308;类似于中间体a。
1868、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物309;类似于化合物238。
1869、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物310;类似于化合物285。
1870、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物311;类似于化合物285。
1871、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物312;类似于化合物244。
1872、使用在方案28和29中描述的合成途径从适当的起始原料制备化合物313;类似于化合物244。
1873、使用在方案28和29描述的合成路线从适当的起始原料制备化合物315;类似化合物314。
1874、使用在方案28和29中描述的合成路线从适当的起始原料制备化合物316;类似化合物238。
1875、使用在方案28和29中描述的合成路线从适当的起始原料制备化合物317;类似化合物238。
1876、化合物319的合成
1877、
1878、步骤1:将中间体b(0.04g,0.12mmol)溶于无水dmf(1.0ml)。将hatu(0.088g,0.23mmol)和三乙胺(0.048ml,0.35mmol)加入反应混合物,随后是(s)-2-氨基-2-(2-氟苯基)-乙-1-醇(0.044g,0.23mmol)。在室温下搅拌反应混合物16小时。lcms显示产品形成m/z483.0和少量原料。在室温下再加入0.044g,(0.23mmol)的(s)-2-氨基-2-(2-氟苯基)-乙-1-醇,将反应混合物搅拌额外4小时。lcms指出产品形成m/z 483.0。反应混合物用去离子水稀释,用dcm(10.0ml x 3)萃取。经合并的dcm层用盐水洗涤,分开和在无水na2so4上干燥。蒸发有机层,提供粗制产品。
1879、c23h20clfn6o3[m+h]+的m/z计算值:483;得到:483.0
1880、步骤2:将上述酰胺(0.06g,0.12mmol)溶于无水dcm(2.5ml)。加入0.03ml(0.23mmol)dast,反应混合物在0℃温度搅拌2小时。lcms指出产品形成m/z 465.2。在0℃加入固体k2co3(0.06g,0.46mmol),反应混合物逐渐温热至室温。反应混合物用nahco3水溶液稀释,用dcm(10.0ml x 3)萃取。有机层用盐水洗涤,分开和在无水na2so4上干燥。蒸发溶剂,提供粗制产品。纯化通过制备型tlc进行,流动相:95:05,dcm:meoh。获得纯产品化合物319(44.2mg),是固体(收率82.2%);c23h18clfn6o3[m+h]+的m/z计算值:465;得到:465.2。
1881、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物320;类似化合物319。
1882、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物321;类似化合物319。
1883、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物322;类似化合物319。
1884、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物325;类似化合物320。
1885、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物326;类似化合物320。
1886、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物330;类似化合物319。
1887、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物331;类似化合物319。
1888、使用在方案28和30中描述的合成路线从适当的起始原料制备化合物333;类似化合物319。
1889、使用在方案28和30中描述的合成路线从适当的起始原料(手性中心衍生自(2r)-2-氨基-2-苯基乙-1-醇)制备化合物340;类似化合物319。
1890、使用在方案28和30中描述的合成路线从适当的起始原料(手性中心衍生自(2s)-2-氨基-2-苯基乙-1-醇)制备化合物343;类似化合物319。
1891、化合物323的合成:
1892、
1893、将化合物319(0.018g,0.04mmol)溶于甲苯(2.0ml)。加入ddq(0.011g,0.05mmol),反应混合物在50℃搅拌3小时。lcms指出原料m/z 465.2和产品m/z 463.2,~1:0.9比率。反应混合物在65℃搅拌2小时,lcms显示原料m/z 465.2和产品m/z 463.2,1:1.6比率。加入额外的ddq(0.003g,0.012mmol),反应混合物在75℃搅拌5小时。lcms指出反应完成。减压浓缩反应混合物。纯化通过制备型tlc进行,流动相:80:20,etoac:己烷。获得~5.8mg化合物323;c23h16clfn6o2[m+h]+的m/z计算值:463.2;得到:463.2
1894、使用合成化合物323所显示的相同条件从化合物320制备化合物324。
1895、化合物305和306的合成
1896、方案31
1897、
1898、将化合物288(0.015克,0.042mmol)溶解在无水thf(3.0ml)中。在-78℃温度加入0.003ml,0.05mmol的碘甲烷,随后加入0.05ml,0.05mmol的1.0m lda溶液。将反应混合物在-78℃搅拌,并在室温逐渐温热。lcms指示产物形成m/z 368(主要的),未反应的起始原料m/z 354和二甲基化的未知产物m/z 382.1。将反应混合物用饱和的nh4cl溶液猝灭,并用etoac萃取。将有机层干燥并浓缩。通过制备型tlc板(流动相:etoac:己烷75:25v/v ml)执行粗制反应混合物的纯化以分离三个带。通过ms发现,第1个带证实了起始原料的m/z 354,第2个带证实了单甲基取代的产物化合物305的m/z 368,且第3个带证实了二甲基取代的产物化合物306的m/z 382.1。1h nmr(cdcl3)数据证实了在咪唑环上的单甲基取代。注:1h nmr数据证实了产物形成和纯产物分离。
1899、与方案18a中的化合物129类似地制备化合物216。ms:[m+1]=395。
1900、与方案18a中的化合物129类似地制备化合物217。ms:[m+1]=381。
1901、方案32
1902、
1903、化合物218的合成:
1904、
1905、在0℃向来自方案27的5-(乙氧基羰基)-16-甲氧基-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-甲酸(0.609g,1.65mmol)在dmf(10ml)中的搅拌溶液中加入nahco3(0.749g,8.9mmol)和nbs(0.793g,4.45mmol)。使该反应进行至环境温度过夜。然后将该反应物用etoac稀释,冷却至0℃,并在搅拌下小心地加入饱和硫代硫酸钠。起泡停止以后,将有机层分离,用饱和nahco3,盐水洗涤,并经mgso4干燥。过滤并除去溶剂,得到粗制溴化物,将其通过使用0-80%的etoac在己烷类中的溶液梯度洗脱的快速柱色谱法纯化。得到424.2mg(64%)微黄色固体。ms:[m+1]=405。
1906、向在厚壁rbf中的来自上面的溴化物(286.7mg,0.709mmol)中加入cui(121.5mg,0.638mmol),三甲基甲硅烷基乙炔(1.04g,10.7mmol),三乙胺(0.717g,7.09mmol),二环己基(2’,6’-二甲氧基联苯-2-基)膦(0.349g,0.851mmol)和1,4-二噁烷(2.5ml;脱气)。将反应容器用氮气净化,并加入双(三苯基膦)二氯化钯(ii)(298.2mg,0.425mmol)。将反应混合物在室温搅拌30分钟,然后在密闭试管条件下在100℃加热16小时,用etoac稀释,并用饱和nahco3,盐水洗涤,并干燥(mgso4)。将过滤并浓缩的反应混合物使用0-100%的etoac在己烷类中的溶液梯度进行硅胶柱色谱法,得到157.9mg(53%)作为淡棕色固体的期望的三甲基甲硅烷基乙炔产物。ms:[m+1]=422。
1907、将上面得到的三甲基甲硅烷基炔烃(128.7mg,0.305mmol)用在thf(0.9ml),水(0.75ml)和meoh(0.15ml)的溶剂混合物中的氢氧化锂(36.6mg,1.53mmol)在室温处理2小时。然后将混合物用稀盐酸酸化至ph 3-4,并用etoac(3×)萃取。发现在水层中的残余沉淀物是产物,并通过过滤收集,并与从有机层分离的产物合并以得到95.6mg作为微黄色固体的酸。
1908、向在thf(1.3ml)和二氯甲烷(1.3ml)中的该酸(95.6mg,0.298mmol)中加入n,o-二甲氧基胺盐酸盐(232.4mg,2.38mmol),edc盐酸盐(456.7mg,2.38mmol),hobt水合物(91.2mg)和三乙胺(0.833ml,5.93mmol)。搅拌16小时后,将该反应物用etoac稀释,并用饱和nh4cl洗涤。将水层分离,并用etoac(3×)萃取,将合并的有机层用饱和nahco3,盐水洗涤并干燥(mgso4)。过滤随后除去溶剂,得到104.8mg作为微黄色固体的酰胺。
1909、向在冰-盐浴中冷却的来自上面的weinreb酰胺(20.1mg,0.0552mmol)在无水thf(0.8ml)中的搅拌溶液中缓慢地加入4-氟苯基溴化镁溶液(1m thf;0.828ml)。将反应混合物历时4小时搅拌至环境温度,然后用饱和nh4cl猝灭,用etoac(3×)萃取,用饱和nahco3,盐水洗涤,并干燥(mgso4)。将过滤并浓缩的混合物使用5%的meoh在dcm中的溶液进行制备型tlc,得到2.0mg作为灰白色固体的化合物218。ms:[m+1]=400。
1910、与在方案32中描述的化合物218类似地制备化合物219。ms:[m+1]=416。
1911、化合物220的合成:
1912、
1913、将5-苯甲酰基-9-乙炔基-16-甲氧基-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯(90.3mg,0.237mmol;与218类似地得到)在室温在thf(1.5ml)中搅拌。加入nabh4(26.8mg,0.71mmol)。1小时以后,将反应物用nh4cl猝灭5分钟,并用etoac萃取。将有机层分离,并用盐水洗涤和经mgso4干燥。过滤并在真空中除去溶剂,得到澄清的粘稠油,将其用三乙基硅烷(241.9mg,2.08mmol)和三氟乙酸(0.32ml)在dcm(1.5ml)中的溶液处理3小时。将反应混合物放在rotovap上除去溶剂,用etoac稀释,并用饱和nahco3洗涤。将水层分离,并用etoac萃取,将合并的有机层用盐水洗涤,并经mgso4干燥。将过滤的浓缩物使用2%的meoh在dcm/etoac(1:1)中的溶液进行制备型tlc,得到2.5mg作为澄清膜样固体的化合物220。ms:[m+1]=370。
1914、与在方案32中描述的化合物220类似地制备化合物221。ms:[m+1]=384。
1915、化合物344的合成:
1916、
1917、将二噁烷(0.5ml)中的方案32的溴化物中间体(26.5mg,0.0656mmol),2-(三正丁基甲锡烷基)-噁唑(58.7mg,0.164mmol)和二氯1,1’-二(二苯基膦基)二茂铁钯(ii)二氯甲烷加合物(7.2mg)在大气压氮下在150℃加热5小时。在冷却后,反应混合物用乙酸乙酯稀释,用饱和nahco3、盐水洗涤,在mgso4上干燥。制备型tlc分离用5% meoh/乙酸乙酯作为洗脱液,提供6.4mg(25%)的噁唑乙基酯产品,是淡黄色固体。
1918、将上述乙基酯(6.4mg)在thf/h2o/meoh溶剂混合物(6:5:1;0.24ml)中用氢氧化锂(6.0mg)处理16小时。加入乙酸(3ml),反应在120℃加热4小时。在冷却后,反应混合物用etoac稀释,用饱和nahco3洗涤。分开水层和用etoac(3x)萃取。经合并的有机溶液用盐水洗涤和在mgso4上干燥。制备型tlc用8% meoh/dcm作为洗脱溶剂系统,提供1.6mg(31%,两步)的脱羧产品化合物344,是淡黄色固体。ms:[m+1]=321。
1919、化合物347的合成:
1920、
1921、在0℃将方案32中的噁唑stille偶联产品(21.2mg,0.054mmol)用硼氢化钠(0.44ml,0.5m,2-甲氧基乙基醚中)处理3天,然后用丙酮(2ml)淬灭60分钟。反应混合物用etoac稀释,用饱和nahco3、盐水洗涤,在mgso4上干燥。在过滤和溶剂除去之后获得粗制醇,在85℃用三溴氧化磷(55mg)/乙腈(1ml)处理7小时。在冷却后,反应用etoac稀释,用饱和nahco3、盐水洗涤,在mgso4上干燥。过滤和减压除去溶剂,提供粗制溴化物产品,是绿色固体。
1922、将上述溴化物在etoac/meoh(5ml,1:1v/v)中用催化量10% pd/炭氢化2小时。过滤通过c盐,用10% meoh/dcm洗涤,氢化后减压除去溶剂提供所希望的甲基产品,其通过制备型tlc(8% meoh/dcm作为洗脱液)纯化,提供3.6mg(20%,三步)化合物347,是淡黄色固体。ms:[m+1]=335。
1923、方案33
1924、
1925、化合物222的合成:
1926、
1927、在室温将氰基酯(407.1mg,1.16mmol)用氢氧化锂(83.5mg,3.49mmol)在thf(6ml),水(5ml)和meoh(1ml)中的溶剂混合物中处理16小时,然后在真空中浓缩,用稀hcl酸化至ph 3-4,并在0℃冷却。将沉淀物通过过滤收集,用少量水洗涤,并干燥以得到271.9mg(73%)作为淡灰色固体的酸。在0℃将该酸(271.9mg)悬浮于thf(2ml)中并搅拌,向其中逐滴加入硼烷二甲硫醚溶液(2m thf;8.4ml)。使该反应进行至环境温度过夜,在冰浴中冷却,用meoh(10ml)猝灭2小时,并在真空中浓缩。将得到的固体残余物在dcm和饱和nahco3之间分配并搅拌20分钟。将水层分离,并用dcm(3×)萃取。将合并的有机层用盐水洗涤并经mgso4干燥。过滤并除去溶剂,得到137.8mg作为微黄色蜡状固体的粗制醇产物。
1928、在100℃将来自上面的醇(137.8mg)用三溴氧化磷(256.3mg,0.894mmol)在1,4-二噁烷(5ml)中处理3小时。在冰浴中冷却后,将反应混合物在搅拌条件下用饱和nahco3(15ml)和etoac(15ml)处理约20分钟。将碱性水层分离,并用etoac(2×)萃取。将合并的有机层用盐水洗涤并经mgso4干燥。过滤并在真空中除去溶剂,得到作为固体糊状物的粗制主要溴化物,将其冷藏保存,且在需要时不经进一步纯化地使用。
1929、在室温将来自上面的粗制溴化物(27.0mg,0.0727mmol)用4-氟苯酚(65.2mg,0.585mmol)和碳酸铯(47.4mg,0.145mmol)处理16小时。将该反应混合物用etoac稀释,用盐水洗涤,并经mgso4干燥。将过滤的浓缩物使用5%的meoh在dcm/etoac(1:1)中的溶液进行制备型tlc,得到1.2mg作为微黄色固体的化合物222。ms:[m+1]=403。
1930、与在方案33中描述的化合物222类似地制备化合物223。ms:[m+1]=403。
1931、与在方案33中描述的化合物222类似地制备化合物224。ms:[m+1]=385。
1932、与在方案33中描述的化合物222类似地制备化合物225。ms:[m+1]=464。
1933、
1934、在70℃将1-(5-氯-2-硝基苯基)-5-(2-乙氧基-2-氧代乙基)-1h-1,2,3-三唑-4-甲酸乙酯(21.2g;与方案11中的14类似地得到)用在etoac/etoh(1:2,300ml)的混合物溶剂中的氯化锡(ii)水合物(60g)处理3小时。加入hcl(40ml;37%),并继续加热3天。加入更多的氯化锡(ii)水合物(25g)和hcl(15ml),并继续加热2天。将反应物冷却,在减压下浓缩为淡棕色油,用etoac(250ml)稀释,并用碳酸钠溶液小心地碱化至ph 8-9。将水层分离,并用etoac重复萃取。将合并的有机层用盐水洗涤,并经mgso4干燥。过滤并除去溶剂,随后在meoh中重结晶,得到3.3g(51%)作为微黄色固体的环化的单-酯。ms:[m+1]=307。
1935、异氰基乙酸叔丁酯的制备:
1936、向甘氨酸叔丁酯盐酸盐(10.0g,60mmol)在dcm(200ml)中的悬浮液中加入edc.hcl(14.9g,78mmol)和三乙胺(12.5ml,89.8mmol)。将反应混合物冷却至-50℃,缓慢地加入甲酸(3.4ml,89.8mmol)在dcm(10ml)中的溶液。将反应混合物在-50℃搅拌1小时,然后在4℃搅拌3小时。加入水(150ml)。搅拌30分钟后,将水层分离,并用dcm(3×)萃取。将合并的有机层用盐水洗涤并经mgso4干燥。过滤并在减压下除去溶剂,得到10g(100%)作为澄清粘稠油的甲酰基酰胺。h1nmr(cdcl3)δ8.23(1h,s),6.17(1h,br s),3.98(2h,d,j=5.5hz),和1.48(9h,s)。
1937、向甲酰基酰胺(10.5g,66mmol)在dcm(180ml)中的溶液中加入三乙胺(36.8ml,264mmol)。将溶液在盐-冰浴中冷却,并缓慢地加入pocl3(7.4ml,79.2mmol)。将反应物在冷浴中搅拌1小时。然后将碳酸钠(7.7g,72.6mmol)在水(90ml)中的溶液加入冷反应混合物中。15分钟以后,除去冷浴,并继续在环境温度搅拌1小时。将水层分离,并用dcm(3×)萃取。将合并的有机层用盐水洗涤并经mgso4干燥。过滤并在减压下除去溶剂,得到7.9g(84%)作为深棕色液体的异氰基乙酸叔丁酯。h1nmr(cdcl3)δ4.12(2h,s),和1.51(9h,s)。
1938、
1939、在氮气气氛下将异氰基乙酸叔丁酯(1.51g,10.7mmol)在dmf(43ml)中的溶液冷却至-50℃。加入叔丁醇钾(1.05g,9.4mmol;压碎)。在-50℃搅拌1小时以后,将1,2,4-三唑中间体(2.32g,6.48mmol;与方案11中的化合物20类似地制备)加入得到的淡红色澄清溶液中,并将反应物搅拌至环境温度过夜。加入饱和nahco3(15ml),并将反应混合物用乙醚(5×)萃取,用盐水洗涤,并干燥(mgso4)。将过滤的浓缩物使用0-100%的etoac在己烷类中的溶液梯度进行硅胶色谱法,得到2.5g(89%)作为微黄色固体的咪唑叔丁基酯产物。ms:[m+1-tbu]=374。
1940、将来自上面的咪唑叔丁基酯(1.1g,2.56mmol)用三氟乙酸(13ml)在dcm(13ml)中的溶液处理3小时或直到所有起始叔丁基酯被水解。然后将该反应物在减压下浓缩。通过重复加入和蒸发甲苯,将残余tfa除去。得到作为深棕色粘稠油状物的酸产物,并不经进一步纯化地使用。ms:[m+1]=374。
1941、
1942、在室温将16-氯-9-氰基-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-5-甲酸乙酯(477mg,1.34mmol);与方案27中的9-氰基-16-甲氧基-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-5-甲酸乙酯类似地得到)用氢氧化锂(80.5mg,3.36mmol)在thf(6ml),水(5ml)和meoh(1ml)的溶剂混合物中处理16小时。将该反应物在减压下浓缩,用稀hcl酸化至ph 3-4,并冷却至0℃。将沉淀物通过过滤收集,用少量水洗涤,并进一步干燥以得到396.2mg粗制三唑并羧酸产物,ms:[m+1]=327。
1943、在0℃向来自上面的粗制酸(396.2mg)在无水thf(7ml)中的悬浮液中逐滴加入硼烷二甲硫醚复合物(10.9ml;2m thf)。使该反应进行至环境温度过夜,并冷却至0℃,然后用meoh缓慢地猝灭。搅拌30分钟后,将该反应混合物在真空中浓缩。将得到的浆料用meoh处理,随后将其在真空中除去。将该过程重复几次。然后将得到的残余物用5%的meoh在dcm中的溶液处理,并用饱和nahco3洗涤。将水层用dcm(3×)萃取,将合并的有机层用盐水洗涤并经mgso4干燥。过滤并除去溶剂,得到粗制醇产物([m+1]=313)和对应伯酰胺(由于氰基的水解)([m+1]=331)的混合物。得到388.8mg该粗制混合物,并不经进一步纯化地使用。
1944、在100℃将来自上面的醇混合物(388.8mg)用三溴氧化磷(2.02g)在1,4-二噁烷(10ml)中处理8小时。将该反应物冷却至0℃,并用饱和nahco3(15ml)小心地猝灭。搅拌20分钟以后,将反应混合物用etoac(3×)萃取,用盐水洗涤,并经mgso4干燥。过滤并在减压下除去溶剂,得到作为粘稠糊状物的粗制溴化物,将其不经进一步纯化用于下一步。
1945、
1946、使用从上面制备的溴化物,与方案33中的化合物222类似地制备化合物226。ms:[m+1]=389。
1947、以与在方案34中描述的化合物226类似的方式制备化合物227。ms:[m+1]=407。
1948、以与在方案34中描述的化合物226类似的方式制备化合物228。ms:[m+1]=407。
1949、
1950、化合物229的合成:
1951、与方案32中的苄基化合物220类似地制备在方案35中所示的苄基类似物229。ms:[m+1]=411。
1952、化合物230的合成:
1953、与方案32中的酮218类似地制备在方案35中所示的酮类似物230。ms:[m+1]=474。
1954、化合物231的合成:
1955、与方案32中的苄基化合物220类似地制备在方案35中所示的苄基类似物231。ms:[m+1]=460。
1956、sch方em案e 36
1957、
1958、在室温将化合物63(0.805g,1.78mmol;来自方案18a)用氢氧化锂(0.128g,5.34mmol)在thf(6ml),水(5ml)和meoh(1ml)的溶剂混合物中处理16小时。然后将该反应物在真空中浓缩,用稀hcl酸化至ph 3-4。将得到的沉淀物通过过滤收集,用水洗涤并干燥以得到0.638g作为黄色固体的酸。ms:[m+1]=424。
1959、将来自上面的酸(0.638g,1.5mmol)用nbs(1.61g,9mmol)和nahco3(1.51g,18mmol)在室温处理16小时。将反应混合物冷却至0℃,小心地并缓慢地加入饱和硫代硫酸钠(水溶液)。将其用etoac(2×)萃取,用饱和nahco3,盐水洗涤,并经mgso4干燥。将过滤的浓缩物用0-100%的etoac在己烷类中的溶液梯度进行硅胶色谱法,得到0.580g(72%)作为微黄色固体的二-溴产物。ms:[m+1]=538。
1960、使用上面制备的溴化物,与方案18a中的化合物55类似地制备化合物232。ms:[m+1]=439。
1961、使用上面制备的溴化物,与方案18a中的化合物55类似地制备化合物235。ms:[m+1]=440。
1962、与方案21中的化合物161类似地制备化合物236炔烃部分。ms:[m+1]=384。
1963、与方案21中的化合物161类似地制备化合物241炔烃部分。ms:[m+1]=385。
1964、
1965、化合物247的合成:
1966、将溴化物酯(13.9mg,0.0344mmol)用氢氧化锂(10mg)在thf(0.3ml),水(0.25ml)和meoh(0.05ml)的溶剂混合物中在室温处理16小时。然后将该反应物在真空中浓缩,用稀hcl酸化至ph 3-4,并冷却至0℃。将得到的沉淀物通过过滤收集,用水洗涤并干燥以得到9.5mg(74%)作为浅棕色固体的酸。ms:[m+1]=377。
1967、向在dcm(0.15ml)中搅拌的来自上面的酸(5.1mg,0.0136mmol)中加入草酰氯(8.6mg,0.0678mmol)和dmf(5ul)。搅拌2小时以后,将溶剂和多余的试剂在真空中除去。将得到的残余物再悬浮于dcm(0.15ml)中,在冰-盐浴中冷却,并逐滴加入甲胺的乙醇溶液(100ul;33%)。搅拌20分钟以后,将反应混合物应用于制备型tlc板,并使用5%的meoh在dcm中的溶液作为洗脱液分离产物。得到4.3mg(81%)作为白色固体的化合物247。ms:[m+1]=390。
1968、与在方案32中描述的化合物247类似地制备化合物248。ms:[m+1]=430。
1969、
1970、在0℃向悬浮于dcm(2ml)中的酸(108.0mg,0.335mmol)中缓慢地加入草酰氯(170.1mg,1.34mmol),随后加入dmf(20ul)。起泡停止以后,除去冰浴,并使该反应在室温进行2小时。将溶剂和多余的试剂在真空中除去。将得到的浅棕色固体冷却至0℃。加入nabh4溶液(2.2ml;1.5m在甲氧基乙氧基乙烷中)。30分钟以后,将反应物用1n hcl(0.2ml)猝灭,并继续搅拌直到起泡停止。加入etoac(10ml)和饱和nahco3(10ml),并将其搅拌过夜。将水层分离,并用etoac(3×)萃取;将合并的有机层用盐水洗涤并经mgso4干燥。过滤并除去溶剂,得到97.0mg(94%)作为微黄色固体的醇。ms:[m+1]=309。
1971、将来自上面的醇(97.0mg,0.315mmol)用戴斯-马丁过碘烷(266.9mg,0.629mmol)在dcm(2ml)中处理1小时。将该反应混合物用dcm稀释,用饱和nahco3洗涤。将水层分离,并用dcm(3×)萃取,将合并的有机层用盐水洗涤,并经mgso4干燥。过滤并在减压下除去溶剂,得到定量收率的作为淡棕色固体的粗制醛,将其不经进一步纯化地使用。
1972、如在方案33中描述的,使用来自上面的醛,与方案16中的化合物48类似地制备化合物250。ms:[m+1]=362
1973、如在方案33中描述的,与化合物250类似地制备化合物251。ms:[m+1]=376。
1974、如在方案33中描述的,与化合物250类似地制备化合物252。ms:[m+1]=364。
1975、如在方案33中描述的,与化合物250类似地制备化合物253。ms:[m+1]=452。
1976、方sch案eme3 73
1977、
1978、在120℃将酸(方案15中的16,x=ome;258.1mg,0.941mmol)用乙酸(2ml)处理5小时。然后在真空中除去溶剂。将固体残余物在水(7ml)中用声处理进行处理,过滤,用水洗涤,并干燥以得到158.4mg(73%)作为淡棕色固体的脱羧产物。ms:[m+1]=231。
1979、
1980、以与方案11中的化合物167类似的方式制备化合物257。ms:[m+1]=364。
1981、以与方案11中的化合物167类似的方式制备化合物258。ms:[m+1]=336。
1982、化合物262的合成:
1983、
1984、在盐-冰浴中冷却下在thf(0.5ml)中搅拌苄基三苯基溴化鏻(29.0mg,0.0669mmol)。加入氢化钠(4.12mg,0.103mmol;60%油悬浮液)。搅拌20分钟以后,加入醛(15.8mg,0.0515mmol)。使该反应物历时4小时缓慢地温热至室温,然后用饱和nh4cl猝灭,用etoac(3×)萃取,用盐水洗涤,并经mgso4干燥。将化合物262通过重复的制备型tlc(使用2%的meoh在dcm中的溶液)分离。分离出1.1mg白色固体。ms:[m+1]=381。
1985、
1986、在室温将起始酯(76.4mg,0.235mmol)用n-溴琥珀酰胺(83.6mg,0.470mmol)在乙腈(2.3ml)中处理3天。向反应混合物中加入饱和硫代硫酸钠。搅拌15分钟以后,将水层分离,并用etoac(2×)萃取。将合并的有机层用盐水洗涤并经mgso4干燥。将溴化物产物通过使用己烷类:etoac=1:3作为洗脱溶剂的制备型tlc分离。得到50.2mg(52%)浅棕色泡沫状固体。ms:[m+1]=405。
1987、在氮气大气压下向来自上面的溴化物(24.1mg,0.0596mmol)中加入苯基硼酸(10.3mg,0.083mmol),四(三苯基膦)钯(0)(6.9mg,0.006mmol),二甲氧基乙烷(0.69ml;脱气)和na2co3水溶液(77ul;2m)。将反应物在100℃加热5小时,冷却至室温,用etoac稀释,用饱和nahco3,盐水洗涤,并经mgso4干燥。用己烷类:etoac=1:3进行制备型tlc,得到17.2mg(72%)作为微黄色无定形物的suzuki偶联产物。ms:[m+1]=402。
1988、化合物272,273和277的合成:
1989、从上面的咪唑酯开始,与方案11中的化合物167类似地制备化合物272。ms:[m+1]=440。
1990、从上面的咪唑酯开始,与方案11中的化合物167类似地制备化合物273。ms:[m+1]=412。
1991、与方案11中的化合物167类似地制备化合物277。ms:[m+1]=378。
1992、以与上面详述类似的方式(参见方案37),通过suzuki偶联来制备化合物279。ms:[m+1]=436。
1993、化合物318的合成
1994、步骤1.在dme(0.55ml)和tfa(0.122ml)中,将方案37中制备的溴化物原料5-溴-16-甲氧基-2,3,4,10,12五氮杂四环-[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-甲酸酯(35.4mg,0.088mmol)用cui(13.3mg,0.07mmol),x-phos(35.9mg,0.088mmol)处理。烧瓶用n2冲洗,向该混合物加入cl2pd(pph3)2(30.7mg,0.0438mmol)。反应加热至100c持续20h,冷却,在etoac与水之间分配。干燥有机相并浓缩,提供粗制产品以及脱硅烷化的物质,5-乙炔基-16-甲氧基-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-甲酸乙酯。两种化合物通过制备型tlc(hex/etoac 1:2)分离。
1995、步骤2.将来自步骤1的5-乙炔基-16-甲氧基-2,3,4,10,12-五氮杂四环-[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-甲酸乙酯(6.2mg,0.0177mmol)溶于dmf(0.2ml)。向该溶液加入碘苯(10.9mg,0.0532mmol),tea(12.5ul),cui(0.7mg,0.0035mmol),和pd(pph3)4(10.2mg,0.009mmol)。混合物在rt下搅拌过夜。混合物用水稀释,用etoac萃取3x。有机相用盐水洗涤,干燥。浓缩提供9.7mg纯化合物318。
1996、
1997、化合物280的合成:
1998、在氮气气氛下向化合物267(11.7mg,0.0274mmol)中加入二环己基[2-(2,4,6-三异丙基苯基)苯基]磷烷(phosphane)(7.8mg,0.0164mmol),碳酸铯(22.3mg,0.0685mmol)和乙腈(0.30ml)。将反应烧瓶用氮气净化,并加入二氯双(乙腈)钯(ii)(1.42mg,0.0055mol)。在室温搅拌30分钟以后,加入三甲基甲硅烷基乙炔(80.7mg,0.822mmol),并将反应物在90℃加热5小时,冷却至室温,用etoac稀释,并用饱和nahco3洗涤。将水层分离,并用etoac(2×)萃取,将合并的有机层用盐水洗涤并经mgso4干燥。将过滤的浓缩物使用5%的meoh在dcm/etoac(1:1)中的溶液进行制备型tlc,得到4.1mg作为微黄色固体的三甲基甲硅烷基乙炔衍生物。ms:[m+1]=489。
1999、在室温将来自上面的三甲基甲硅烷基乙炔(4.1mg,0.0084mmol)用碳酸钾(1.2mg,0.0084mmol)在甲醇(0.2ml)中处理3小时。使用7%的meoh在dcm/etoac(1:1)中的溶液作为洗脱溶剂进行制备型tlc,得到1.6mg作为微黄色固体的化合物280。ms:[m+1]=417。
2000、化合物284,301和302的合成:
2001、从化合物240开始与化合物280类似地制备化合物284。ms:[m+1]=403。
2002、从化合物264开始与化合物280类似地制备化合物301。ms:[m+1]=437。
2003、从化合物245开始与化合物280类似地制备化合物302。ms:[m+1]=435。
2004、
2005、化合物289,290,291和292的合成:
2006、与在方案30中描述的化合物263类似地制备化合物289。ms:[m+1]=399。
2007、与在方案30中描述的化合物263类似地制备化合物290。ms:[m+1]=399。
2008、与在方案29中描述的化合物243类似地制备化合物291。ms:[m+1]=337。
2009、与在方案29中描述的化合物243类似地制备化合物292。ms:[m+1]=337。
2010、
2011、化合物298的合成:
2012、在0℃将在thf(2.4ml)中的酯(107.9mg,0.264mmol)用硼氢化锂溶液(0.264ml;2mthf)处理。使该反应物历时4小时温热至环境温度,然后用饱和nahco3缓慢地猝灭,用etoac(4×)萃取,用盐水洗涤,并经mgso4干燥。过滤并除去溶剂,得到77.3mg(86%)作为微黄色固体的醇。
2013、在95℃将来自上面的醇(16.4mg,0.0448mmol)用三溴氧化磷(25.7mg,0.0895mmol)在1,4-二噁烷(0.5ml)中处理3小时。然后将该反应物冷却至0℃,用饱和nahco3(5ml)猝灭20分钟,并用etoac(3×)萃取,用盐水洗涤,并经mgso4干燥。过滤并干燥,得到16.6mg微黄色固体,将其溶解在无水meoh(18ul)和thf(0.35ml)中。将其冷却至0℃,并加入nah(9.2mg;60%悬浮液)。在0℃搅拌2小时以后,将反应物用饱和nahco3猝灭,用etoac(3×)萃取,用盐水洗涤,并经mgso4干燥。使用10%的meoh在dcm中的溶液进行制备型tlc,得到0.8mg作为微黄色固体的化合物298。ms:[m+1]=381。
2014、
2015、如早前所述(参见方案21),将起始醇(616mg)转化成对应的溴化物。将得到的粗制溴化物溶解在无水甲醇(23ml)中,并冷却至0℃。逐份加入nah(932mg;60%悬浮液)。起泡停止以后,将反应混合物加热至回流保持30分钟,然后冷却至室温,并用2n hcl(11ml)处理。将得到的沉淀物通过过滤收集,并将期望的甲基醚通过硅胶色谱法(使用0-10%的meoh在dcm中的溶液梯度洗脱)分离。收集217mg微黄色固体。ms:[m+1]=279。
2016、
2017、化合物299和300的合成
2018、使用上面的甲基醚中间体,与化合物289类似地制备化合物299。ms:[m+1]=461。
2019、使用上面的甲基醚中间体,与化合物289类似地制备化合物300。ms:[m+1]=385。
2020、方案38
2021、
2022、化合物327的合成:
2023、
2024、步骤1.中间体b的异丙氧基类似物(15-氯-9-[(丙-2-基氧基)甲基]-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-羧酸)用如方案28中所示的相同反应程序以类似的方式制备。将该化合物(0.05g,0.13mmol)溶于无水dmf(2.5ml)。将hatu(0.061g,0.16mmol)和氨基乙醛二甲基缩醛(0.029ml,0.27mmol)加至反应混合物,随后是0.047ml(0.26mmol)二异丙基乙胺。在室温下搅拌反应混合物4小时。lcms显示产品形成m/z 461.3和少量原料。将额外的0.030g,(0.08mmol)hatu和0.029ml(0.27mmol)氨基乙醛二甲基缩醛加至反应混合物,搅拌反应混合物额外2小时。lcms指出反应完成。反应混合物用去离子水稀释,用乙酸乙酯(15.0ml x 3)萃取。经合并的乙酸乙酯层用盐水洗涤,分开和在无水na2so4上干燥。蒸发有机层,提供粗制产品15-氯-n-(2,2-二甲氧基乙基)-9-[(丙-2-基氧基)甲基]-2,4,8,10,11-五氮杂四环-[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酰胺(62mg,100%);c21h25cln6o4[m+h]+的m/z计算值:461;得到:461.3。
2025、步骤2.除去缩醛部分:将thf(2.0ml)中的上述化合物(0.062g,0.13mmol)与1.3ml,(1.3mmol)的1.0m hcl溶液在35-40℃温度搅拌16小时。lcms显示脱保护的醛m/z415.3。反应混合物用乙酸乙酯30.0ml稀释,用nahco3饱和溶液、随后盐水洗涤。分开有机层,在无水na2so4上干燥。蒸发溶剂,提供粗制产品(15-氯-n-(2-氧代乙基)-9-[(丙-2-基氧基)甲基]-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲酰胺(55mg,100%);c19h19cln6o3[m+h]+的m/z计算值415,得到415.3。
2026、步骤3.将上述醛(0.055g,0.13mmol)溶于无水thf(5.0ml)。加入burgess试剂(0.064g,0.26mmol),反应混合物在70℃加热2小时。lcms显示原料m/z 415和产品m/z397.2的混合物。再加入额外的burgess试剂(0.032g,0.13mmol),反应混合物在70℃加热3小时。lcms显示产品形成m/z 397.2。反应混合物用乙酸乙酯(30.0ml)稀释,用nahco3饱和溶液、随后盐水洗涤。分开有机层,在无水na2so4上干燥。蒸发溶剂,提供粗制产品。粗制产品的纯化通过制备型tlc板进行:流动相:etoac:meoh,96:04v/v ml。获得13.5mg固体化合物327(25.4%收率);c19h17cln6o3[m+h]+的m/z计算值397,得到397.2。
2027、化合物341与化合物327类似地制备,如方案38中所示。
2028、化合物349的合成:
2029、
2030、与将中间体a转化为中间体b类似地,将中间体c(方案28中制备)转化为相应的羧酸(15-氯-9-(苯氧基甲基)-2,4,8,10,11五氮杂-四环[11.4.0.02,6.08,12十七烷-1(17),3,5,9,11,13,15-七烯-5-羧酸。然后与示于方案38的化合物327类似地以三步程序用适当的试剂将该化合物转化为化合物349。
2031、化合物350的合成:
2032、
2033、化合物350以对化合物349显示的类似方式合成,使用适当的原料和方案38中描述的相同反应。
2034、化合物355的合成:
2035、
2036、化合物355以对化合物349显示的类似方式合成,使用适当的原料和方案38中描述的相同反应。
2037、方案39
2038、
2039、化合物342的合成:
2040、
2041、制备r=ch2cf3的上述醛(15-氯-9-[(2,2,2-三氟乙氧基)甲基]-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛),其类似于方案29所示的醛(15-氯-9-(甲氧基甲基)-2,4,8,10,11五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛)。将该化合物(0.04g,0.1mmol)溶于3.0ml无水meoh,在室温下滴加k2co3(0.028g,0.2mmol)。加入ohira bestman试剂(0.02ml,0.14mmol),反应混合物在室温下搅拌16小时。lcms显示产品形成m/z394.2。减压浓缩反应混合物,用20.0ml碳酸氢钠水溶液稀释。过滤ppts,用去离子水洗涤,在干燥之后获得34.0mg固体(收率85.9%);c17h11clf3n5o[m+h]+的m/z计算值:394;得到:394。
2042、化合物351的合成:
2043、
2044、制备r=ch2ph的上述醛9-[(苄氧基)甲基]-15-氯-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛,其类似于方案29所示的醛(15-氯-9-(甲氧基甲基)-2,4,8,10,11五-氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛)。与化合物342类似地,将其转化为化合物351,如方案39中所示。
2045、化合物353的合成:
2046、上述乙炔衍生物化合物353(15-氯-5-乙炔基-9-(2-氟苯氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯)制备自12-氯-9-氧代-2,4,8-三氮杂三环[8.4.0.02,6]十四-1(10),3,5,11,13-五烯-5-甲酸乙酯和2-(2-氟苯氧基)乙酰肼,使用与方案28描述的那些类似的程序。
2047、化合物328的合成:
2048、制备醛(15-甲氧基-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环-[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛),其类似与方案29对15-氯-9-(甲氧基甲基)-2,4,8,10,11五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯-5-甲醛的描述。与如方案39中对化合物342所示类似地,将该化合物转化为化合物328,提供5-乙炔基-15-甲氧基-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,9,11,13,15-七烯(化合物328)。
2049、化合物352和354的合成:
2050、以与方案29对化合物339所报告类似的方式,化合物352和354分别制备自化合物351和353。
2051、方案40
2052、
2053、化合物329的合成:
2054、步骤1:于0c在thf(1.5ml)中搅拌化合物103(描述于方案18a)(107mg,0.29mmol),向其加入dibal(0.73ml,1m溶胶,在己烷中,0.73mmol)。搅拌混合物2小时。加入额外0.5当量的dibal以推动反应完成。在30分钟之后,混合物用饱和nahco3淬灭,用etoac(3x)萃取。有机相用盐水洗涤和干燥(mgso4)。浓缩提供粗制产品,其通过制备型tlc(10% meoh/dcm)纯化,提供57mg(61%)的[15-甲氧基-9-(甲氧基甲基)-2,4,8,10,11-五氮杂四环[11.4.0.02,6.08,12]十七-1(17),3,5,9,11,13,15-七烯-5-基]甲醇。
2055、步骤2:在accn(1ml)中搅拌上述醇(18.9mg,0.0577mmol),向其加入pobr3(50mg,0.173mmol)。于90c搅拌混合物3小时,冷却,用etoac稀释,用饱和nahco3处理。混合物用etoac萃取。合并有机相,用盐水洗涤,干燥,浓缩,提供粗制产品,其直接用于后续反应中。
2056、步骤3:在充h2气球下将上述粗制溴化物与10% pd/c(催化量)在8ml的1:1meoh/etoac中搅拌48小时。混合物过滤通过c盐。浓缩提供粗制产品,将其通过制备型tlc(15%meoh/dcm)纯化,提供11.3mg化合物329,是白色固体。
2057、化合物356的合成:
2058、
2059、方案41
2060、
2061、在thf(0.6ml)、水(0.5ml)和meoh(0.1ml)的溶剂混合物中,将9-叔丁基5-乙基16-氯-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-5,9-二羧酸酯(方案34中制备)(100.7mg,0.234mmol)用氢氧化锂(28.1mg,1.17mmol)处理12小时。反应混合物然后减压浓缩除去绝大多数有机溶剂,再悬浮于乙酸(3ml),在120℃加热20小时。所得褐色透明溶液然后滴加入30ml搅拌的冷水。溶液然后在冰浴中冷却超过30min。过滤收集所得沉淀,用水洗涤,再干燥,提供45.4mg(64%,两步)的16-氯-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-羧酸,是褐色固体。ms:[m+1]=302。
2062、向dmf(0.5ml)中的上述一元酸(45.3mg,0.150mmol)加入n,o-二甲基盐酸羟胺(22.0mg,0.225mmol),hatu(62.7mg,0.165mmol),和n,n-二异丙基乙胺(58.2mg,0.450mmol)。在搅拌2小时之后,反应混合物用etoac稀释,用0.5n hcl、饱和nahco3盐水洗涤,在mgso4上干燥。随后减压过滤除去溶剂在真空中,提供40.8mg(79%)weinreb酰胺,是淡黄色固体。ms:[m+1]=345。
2063、在-78℃向无水thf(0.5ml)中的上述酰胺(40.8mg,0.118mmol)加入二异丁基铝氢化物溶液(0.5ml;1m,在己烷中)。在1h搅拌之后,让反应缓慢温热至-10℃,冷却返回-78℃。加入酒石酸钠钾(5ml)的饱和水溶液,搅拌60min。溶液用etoac(4x)萃取,用盐水洗涤,在mgso4上干燥。过滤和除去溶剂,提供18.7mg(55%)的16-氯-2,3,4,10,12-五氮杂四环[11.4.0.02,6.08,12]十七烷-1(17),3,5,8,10,13,15-七烯-9-甲醛,是淡黄色固体。ms:[m+1]=286。
2064、向于rt在meoh(1ml)中搅拌的上述醛(18.7mg,0.0655mmol)加入碳酸钾(18.1mg,0.131mmol)和二甲基1-重氮基-2-氧代丙基膦酸酯(21.4mg,0.111mmol)。在16hr搅拌之后,反应用etoac稀释,用nahco3饱和水溶液洗涤。分层,用etoac萃取两次;经合并的有机溶液用盐水洗涤,在mgso4上干燥。过滤和除去溶剂,提供粗制混合物,自其分离所希望的炔化合物356:使用制备型tlc,5% meoh/etoac/二氯甲烷(1:1)作为洗脱液。获得8.7mg化合物356,是浅黄色固体。ms:[m+1]=282。
2065、使用适当的原料制备化合物357-375,382,383,385-398,399-412,414,416和425-428,其方式类似方案29,30和34中描述的那些。对于化合物398,在sonogashira反应中使用三氟碘乙烷。
2066、化合物376-381如方案21中所示在引入噁唑啉部分之前制备。
2067、化合物384与化合物280类似地在引入噁唑啉部分之后制备。
2068、化合物413如方案39中所示制备,用苄基溴代替碘甲烷。
2069、化合物415与化合物280类似地制备,开始于化合物452和3-甲基-1-丁炔。在该反应混合物中形成两种额外的化合物-化合物453(二-炔产品)和化合物454(环cl原子用炔部分替换)。
2070、化合物417-423如方案39中所示制备,用适当取代的苄基溴代替碘甲烷。
2071、化合物424与化合物280类似地制备,用溴化物作为原料。
2072、化合物429通过在苯酚与咪唑甲酰基甲醇之间的mitsunobu反应制备(参见方案15和随后实施例的反应条件)。
2073、化合物430如方案29中所示制备,用bnbr将咪唑甲酰基甲醇烷基化,将nah/thf用作碱性介质。
2074、化合物431-432如方案29中所示制备,用适当取代的苄基溴将咪唑甲酰基甲醇烷基化,将nah/thf用作碱性介质。
2075、化合物433-435类似化合物429制备,使用适当取代的苯酚。
2076、化合物436类似化合物430制备,在相同的碱性条件下用2-f苄基溴将适当的初始醇烷基化。
2077、化合物437类似化合物430制备,在相同的碱性条件下用3-f苄基溴将适当的初始醇烷基化。
2078、化合物438类似化合物430制备,在相同的碱性条件下用3-cl苄基溴将适当的初始醇烷基化。
2079、化合物439类似化合物430制备,在相同的碱性条件下用2-cl苄基溴将适当的初始醇烷基化。
2080、化合物440-443类似化合物429通过在苯酚与相应伯醇之间的mitsunobu反应制备。
2081、化合物444-445类似化合物274和215如方案29中所示制备,用适当取代的苄基溴将咪唑甲酰基甲醇烷基化,将nah/thf用作碱性介质。
2082、化合物446-447:如方案39中所示开始于相似的醛,这些化合物用如方案33中所示的烯化条件类似地制备。
2083、化合物448的制备开始于化合物356且类似于方案29的条件,sonogashira反应在pdcl2(pph3)2催化下进行,用苯甲酰氯代替ph-i。
2084、化合物449的制备开始于化合物403,用bnbr作为烷基化剂和nah/thf作为碱性条件进行苄基化,视需要加热。
2085、化合物450的制备开始于化合物403,用3-f-bnbr作为烷基化剂和nah/thf作为碱性条件进行苄基化,视需要加热。
2086、化合物451的制备开始于化合物403,用2-f-bnbr作为烷基化剂和nah/thf作为碱性条件进行苄基化,视需要加热。
2087、化合物455的制备开始于化合物256且类似于方案29的条件,sonogashira反应在pdcl2(pph3)2催化下进行,用苯甲酰氯代替ph-i。
2088、化合物452-454在与化合物280相同的条件下合成,开始于化合物452和3-甲基-1-丁炔。
2089、化合物456-471以与方案28和29描述的那些类似的方式制备;使用适当的原料。用nah代替kotbu形成咪唑环。
2090、通过ms和1h nmr表征化合物180-471。ms表征总结在下面表5中。
2091、表5.化合物180-471的ms表征
2092、
2093、
2094、
2095、
2096、
2097、
2098、
2099、
2100、
2101、
2102、
2103、
2104、
2105、
2106、
2107、
2108、
2109、
2110、
2111、
2112、
2113、
2114、
2115、
2116、
2117、
2118、
2119、
2120、
2121、
2122、
2123、
2124、
2125、
2126、
2127、
2128、
2129、
2130、
2131、
2132、
2133、
2134、
2135、
2136、
2137、
2138、
2139、
2140、
2141、
2142、
2143、进行与方案1至37中所示的那些近似和类似的反应,在本技术中也明确预见到以下化合物:
2144、
2145、
2146、
2147、
2148、
2149、
2150、
2151、
2152、
2153、实施例105:评估含有α5的gabaa受体(gabaar)正变构调节剂活性
2154、步骤1:建立gabaar亚基(α5,β3,γ2,α1,α2和α3)的克隆和制备相应的crna:gabaa-rα5,β3,γ2,α1,α2和α3亚基的人克隆得自商业来源(例如origene,http://www.origene.com和genescript,http://www.genescript.com)。将这些克隆工程改造成prc,pcdm,pcdna和pbluescript ksm载体(用于卵母细胞表达)或其它等效表达载体。使用常规转染试剂(例如,fugene,lipofectamine 2000或其它)瞬时转染宿主细胞。
2155、步骤2-爪蟾卵母细胞表达系统中α5β3γ2,α1β3γ2,α2β3γ2和α3β3γ2亚型的功能性gabaar测定:将编码α5,β3,γ2,α1,α2和α3亚基的crna使用t3 mmessage mmachinekit(ambion)在体外转录,并且注射(以α:β:γ=2:2:1的比例或其它经优化的条件)进从非洲爪蟾新鲜制备的卵母细胞中。培养2天后,使用tevc方案(setup)执行来自卵母细胞的gaba-门控的cl-电流(warner instruments,inc.,foster city,ca)。gaba,苯并二氮杂环庚三烯和地西泮用作参照化合物以验证该系统。
2156、步骤3-评价受试化合物对α5β3γ2亚型的正变构调节剂活性并试验当达到ec50=5μm选择性截止值时对α1至α3偶联的β3γ2亚型的脱靶(off-target)活性:在受试化合物存在下用tevc方案测量来自卵母细胞的gaba-门控的cl-电流。在5-点剂量-响应测定中试验每种受试化合物的正变构调节剂活性。受试化合物包括一些参照化合物(α5β3γ2亚型的文献ec50值是在3-10μm的范围)。对每种化合物得到α5β3γ2亚型的ec50。如果α5β3γ2的ec50≤5μm,那么进一步单个地确定其它三种亚型(α1β2γ2,α2β3γ2和α3β3γ2)的ec50,以便试验所述化合物对α5β3γ2亚型超过其它亚型的选择性。
2157、步骤4-评价其它受试化合物对α5β3γ2亚型的活性并试验当达到ec50=0.5μm选择性截止值时的脱靶活性:使用相同策略试验第二批受试化合物,但是具有更低的ec50截止值(0.5μm)。此外,确定每种化合物的α5β3γ2亚型的ec50。仅仅如果含有α5的受体的ec50<0.5μm,那么试验α1至α3偶联的β3γ2亚型。
2158、实施例106:评价化合物对gabaaα5受体的结合和正变构调节剂活性
2159、(a)受试化合物对gabaar的结合活性
2160、组织培养和膜制备:在稳定地表达gabaa受体(α1β1γ2,α2β3γ2,α3β3γ2和α5β3γ2)的ltk细胞(由merck co.,nj,usa提供)上进行所述结合。在5%co2中将细胞在含有10%血清和抗生素的dmem/f12培养基中接种在100mm培养板中,并且允许其生长1-2天。然后如下通过地塞米松诱导gabaar表达:对于含有α5的gabaar,0.5μm进行1天,对于含有α1,α2和α3的gabaar,2μm进行3天。诱导后,如下收集细胞:刮进dulbecco氏磷酸盐缓冲盐水(dpbs,ph 7.4,invitrogen,carlsbad,ca,usa)中,并在150×g离心10分钟。通过再混悬和离心,将沉淀物洗涤两次。将来自至少5个不同制备的细胞沉淀物组合,悬浮于结合测定缓冲液(50mm kh2po4;1mm edta;0.2mkcl,ph 7.4)中,并通过使用branson sonifier 150(g.heinmann,德国)声处理(3-5次,30秒)制备膜。用牛血清白蛋白(sigma aldrich,st.louis,mo,usa)作为标准,使用bca测定(bio-rad labs,reinach,瑞士)确定蛋白质含量。制备等分试样并在-20℃储存用于进一步在结合测定中使用。
2161、配体结合:如下得到饱和结合曲线:将膜与递增浓度(0.01-8nm)的[3h]rol5-1788(flumazepil,75-85ci/mmol,perkinelmer,ma,usa)一起温育,在有10μm地西泮存在下测量非特异性结合。在等于或低于从饱和曲线确定的含有α1,α2,α3和α5的gabaar的kd值的放射性配体浓度,执行受试化合物的[3h]rol5-1788结合的抑制。
2162、所有结合测定在测定缓冲液中在4℃进行1小时。总的测定体积是0.5ml,对于含有α5的gabaar膜而言含0.2mg/ml蛋白质,对于含α1,α2和α3的gabaar膜而言含0.4mg/ml蛋白质。使用24-cell harvestor(brandel,gaithersburg,md,usa)通过穿过gf/b过滤器过滤终止温育,随后用冰冷的测定缓冲液洗涤3次。将过滤器转移至闪烁瓶,加入5ml闪烁液,涡旋混合并保持在黑暗中。第二天,使用闪烁计数器(beckman coulter,brea,ca,usa)得到放射性。一式三份地进行所有测定。
2163、数据分析:使用graphpad prism软件(graphpad software,inc.,ca,usa)获得饱和和抑制曲线。使用cheng-prusoff方程式ki=ic50/(1+s/kd)确定未标记的配体的平衡解离常数(ki值),其中ic50是抑制50%的[3h]配体结合的未标记配体的浓度,s是放射性配体的浓度,且kd是放射性配体的平衡解离常数。使用化合物的log范围(1nm-10μm)确定ki值,其表示为来自一式三份测定的平均值±sd。
2164、(b)受试化合物对α5β2γ2亚型gabaar的正变构调节剂活性
2165、首先使用与以上所示方案基本上类似的方案,针对它们的增加含有gabaa受体(α5β2γ2)的卵母细胞中gaba的ec20浓度的能力在100nm筛选本发明的化合物。
2166、在第1天,将1ng/32nl的gabaaα5β2γ2cdna注射进一个卵母细胞中。在第2天开始试验。注射进所述卵母细胞中的cdna是α,β和γ的混合物,它们的比率是1:1:10(按重量计),并且要注射进一个卵母细胞中的混合的3个亚基的总重量是在32nl体积中的1ng。也可以在第3天试验被注射的卵母细胞。在这样的情况下,注射进所述卵母细胞中的cdna的量应当减少20%。
2167、使用以下程序试验本发明的化合物。
2168、gaba剂量-响应
2169、1).将8个卵母细胞放在opusxpress的8个腔室中,并用改良的barth氏盐水(mbs)以3ml/min浇盖于其上。使用用3m kcl(0.5-3兆欧姆)回填的玻璃电极。将卵母细胞的膜电位电压钳制在-60mv。
2170、2).将从前述试验中获得的平均ec20 gaba应用5-6次以稳定卵母细胞。在每次gaba应用之间,将卵母细胞用mbs洗涤5-10分钟。
2171、3).进行gaba剂量-效应以获得ec20 gaba值。
2172、对照试验(地西泮或3,5-二苯基哒嗪-4-甲酸甲酯)
2173、1).使用新的卵母细胞来进行新试验。
2174、2).将ec20 gaba应用5-6次以稳定卵母细胞。在每次gaba应用之间,将卵母细胞用mbs洗涤5-10分钟。
2175、3).应用ec20 gaba以获得电流(igaba)。将卵母细胞用mbs洗涤5-10分钟。
2176、4).预先应用1μm地西泮或3,5-二苯基哒嗪-4-甲酸甲酯40秒,随后将1μm地西泮或3,5-二苯基哒嗪-4-甲酸甲酯和ec20 gaba共同应用,以获得i试验。将i试验除以igaba以获得增强(potentiation)(%)。
2177、多剂量的受试化合物
2178、1).重复所述对照试验中的以上步骤1),2)和3)。
2179、2).预先应用第一个浓度的受试化合物40秒,随后将相同浓度的所述受试化合物和ec20 gaba共同应用以获得i试验。将i试验除以igaba以获得增强(%)。
2180、3).抛弃所有试验过的卵母细胞,使用新的卵母细胞,并重复以上步骤1)和2)以试验相同化合物的第二个浓度。每个卵母细胞仅用于单一受试化合物的一个浓度试验。为其它受试化合物重复所述步骤。
2181、在某些实施方案中,本技术的化合物具有小于200nm,小于180nm,小于150nm或小于100nm的对含有α5的gabaar的结合亲和力(由ki表示)。在某些实施方案中,本技术的化合物具有小于50nm的对含有α5的gabaar的结合亲和力(由ki表示)。在某些实施方案中,本技术的化合物具有小于10nm的对含有α5的gabaar的结合亲和力(由ki表示)。
2182、在某些实施方案中,本技术的化合物对含有α5的gabaar的选择性超过含有α1的gabaar。在某些实施方案中,相对于含有α1的gabaar而言,本技术的化合物对含有α5的gabaar的选择性超过50倍,超过100倍,超过500倍或超过1000倍。
2183、在某些实施方案中,本技术的化合物具有小于500nm,小于100nm或小于50nm的对含有α5的gabaar的ec50。在某些实施方案中,本技术的化合物具有小于25nm的对含有α5的gabaar的ec50。
2184、在某些实施方案中,本技术的化合物在100nm使含有α5的gabaar增强超过10%,超过25%,超过50%或超过75%。在某些实施方案中,本技术的化合物在1000nm使含有α5的gabaar增强超过10%,超过25%,超过50%或超过75%。
2185、结合和pam功能活性试验的筛选结果总结在下面表1和2中。
2186、以下表1解释了与本发明的化合物有关的gabaα5结合ki的范围
2187、表1
2188、
2189、
2190、
2191、以下表2解释了与本发明的化合物有关的gabaα5功能增强的范围:
2192、表2
2193、
2194、
2195、本发明的选择的化合物表现出相对于gabaα1,gabaα2或gabaα3的>10倍的gabaα5结合选择性。
2196、本技术的某些化合物表现出相对于gabaα1,gabaα2或gabaα3超过20倍,50倍或100倍的gabaα5结合选择性。
2197、下表6说明本技术化合物相对于gabaα1,gabaα2或gabaα3的gabaα5结合选择性范围:
2198、表6
2199、
2200、
2201、实施例107:3,5-二苯基哒嗪-4-甲酸甲酯在老龄受损(ai)大鼠中的作用
2202、与van niel等人.j.med.chem.48:6004-6011(2005)中的化合物编号6对应的3,5-二苯基哒嗪-4-甲酸甲酯是选择性的含有α5的gabaa r激动剂。其具有+27(ec20)的α5体外效力。使用ram任务研究了3,5-二苯基哒嗪-4-甲酸甲酯在老龄受损大鼠中的作用。此外,还研究了3,5-二苯基哒嗪-4-甲酸甲酯在含有α5的gabaa受体中的受体占有率。
2203、(a)使用辐射臂迷宫(ram)行为任务3,5-二苯基哒嗪-4-甲酸甲酯在老龄受损大鼠中的作用
2204、使用媒介物对照品和4种不同剂量水平的3,5-二苯基哒嗪-4-甲酸甲酯(0.1mg/kg,0.3mg/kg,1mg/kg和3mg/kg,腹膜内),在辐射臂迷宫(ram)行为任务中评估3,5-二苯基哒嗪-4-甲酸甲酯对老龄受损(ai)大鼠的体内空间记忆保留的影响。使用8只ai大鼠执行ram行为任务。对全部8只大鼠试验所有5种治疗条件(媒介物和4种剂量水平)。
2205、所用的ram设备由8个等间距臂组成。升起的迷宫臂(7cm宽度x 75cm长度)从八角形中心平台(30cm直径,51.5cm高度)的每个小平面伸出。臂上的透明侧壁是10cm高且呈65°角以形成波谷。食物孔(4cm直径,2cm深)位于每个臂的远侧端部。froot loopstm(kelloggcompany)用作奖赏。可以放置由plexiglastm(30cm高度x 12cm宽度)构成的块以防止进入任意臂。还提供了围绕设备的众多额外迷宫线索。
2206、首先对ai大鼠进行预先训练试验(chappell等人neuropharmacology 37:481-487,1998)。预先训练试验由习惯期(4天),关于标准赢得-替换任务的训练期(18天)和另一个训练期(14天)组成,在所述另一个训练期中,在实验者指定的臂子集(例如可利用的5个臂和被封闭的3个臂)的呈现和8-臂赢得-替换任务(即,可利用的所有8个臂)的完成之间添加短暂的延迟阶段。
2207、在习惯期中,大鼠熟悉迷宫8-分钟期限,连续4天。在这些期限的每个中,食物奖赏分散于ram上,最初在中心平台和臂上,然后逐步限于臂上。在该习惯期以后,使用标准训练方案,其中食物颗粒位于每个臂的末端。大鼠每天接受一次试验,持续18天。当已经得到全部8个食物颗粒时或当做出16种选择时或15分钟已经过去时,终止每个每日试验。该训练期结束后,进行第二个训练期,其中通过在试验过程中施加短暂延迟来增加记忆要求。在每个试验开始时,封闭8-臂迷宫的3个臂。允许大鼠得到在试验的该最初"信息阶段"中允许进入的5个臂上的食物。然后从迷宫中取出大鼠60秒,在该时间中,除去迷宫上的屏障,由此允许接近所有8个臂。然后将大鼠放回到中心平台上,并允许其在所述试验的该"保留试验"中得到剩余的食物奖赏。封闭的臂的身份和构造在试验中变化。
2208、追踪ai大鼠在保留试验阶段中形成的"错误"的数目。如果大鼠进入已经在试验的延迟前部分中从其取回食物的臂,或者如果大鼠在延迟后期间中重新探访它已经探访过的臂,则在试验中发生错误。
2209、预先训练试验结束后,对大鼠进行在信息阶段(提供一些封闭的臂)与保留试验(提供所有臂)之间具有更延长的延迟间隔(即2-小时延迟)的试验。在延迟期间中,大鼠在它们各自的居住笼中在小车上保持远离试验室内的迷宫的侧面。将ai大鼠在每日试验前30-40分钟用如下5种条件的一次性注射预处理:1)媒介物对照品-5%二甲亚砜,25%聚乙二醇300和70%蒸馏水;2)3,5-二苯基哒嗪-4-甲酸甲酯,0.1mg/kg;3)3,5-二苯基哒嗪-4-甲酸甲酯,0.3mg/kg;4)3,5-二苯基哒嗪-4-甲酸甲酯,1mg/kg);和5)3,5-二苯基哒嗪-4-甲酸甲酯,3mg/kg;通过腹膜内(i.p.)注射。每隔一天施用注射,间隔清除天。在试验阶段内用所有5种条件处理每只ai大鼠。为了抵销任何潜在的偏倚(bia),使用升-降剂量系列评估药物作用,即首先以升序施用剂量系列,并然后以降序重复。因此,每种剂量具有两个测定值。
2210、使用参数统计(配对t检验)对比在不同剂量的3,5-二苯基哒嗪-4-甲酸甲酯和媒介物对照品的背景下ai大鼠在2小时延迟形式的ram任务中的保留试验性能(参见图1)。在试验中发生的错误的平均数对于使用3mg/kg的3,5-二苯基哒嗪-4-甲酸甲酯治疗(错误的平均数±平均值标准误差(sem)=1.31±0.40)明显低于使用媒介物对照品(错误的平均数±sem=3.13±0.62)。相对于媒介物对照品治疗,3,5-二苯基哒嗪-4-甲酸甲酯在3mg/kg显著地改善了记忆性能(t(7)=4.233,p=0.004)。
2211、当用0.3mg/kg tb21007(即含有α5的gabaa r反激动剂)并行地治疗ai大鼠时,3mg/kg的治疗剂量变得无效。使用组合的tb21007/3,5-二苯基哒嗪-4-甲酸甲酯治疗(0.3mg/kg tb21007与3mg/kg 3,5-二苯基哒嗪-4-甲酸甲酯)的大鼠做出的错误的平均数为2.88±1.32,且与用媒介物对照品治疗的大鼠(3.13±1.17平均误差)相差无几。因此,3,5-二苯基哒嗪-4-甲酸甲酯对空间记忆的作用是gabaaα5受体依赖性的作用(参见图1)。
2212、(b)3,5-二苯基哒嗪-4-甲酸甲酯对含有α5的gabaa受体占有率的影响
2213、动物
2214、将成年雄性long evans大鼠(265-295g,charles river,portage,mi,n=4/组)用于gabaaα5受体占有率研究。以12:12光照/黑暗周期将大鼠单个地圈养在通风的不锈钢格栅中。可以随意接近食物和水。在以行为活性剂量评价化合物暴露的另外研究中,将年轻或老龄long evan大鼠(n=2-4/组)用于这些研究。
2215、化合物
2216、将ro 15-4513用作海马和小脑中gabaaα5受体位点的受体占有率(ro)示踪剂。基于它对gabaaα5受体相对于含有其它α亚基的gabaa受体的选择性和因为它已经成功地用于动物和人类中的gabaaα5ro研究,将ro 15-4513选作示踪剂(参见,例如,lingford-hughes等人,j.cereb.blood flow metab.22:878-89(2002);pym等人,br.j.pharmacol.146:817-825(2005);和maeda等人,synapse 47:200-208(2003))。将ro 15-4513(1μg/kg)溶解在25%羟基-丙基β-环糊精中,在ro评价前20分钟静脉内施用。3,5-二苯基哒嗪-4-甲酸甲酯(0.1-10mg/kg)由nox pharmaceuticals(india)合成,将其溶解在25%羟基-丙基β-环糊精中,在示踪剂注射前15分钟静脉内施用。除最高剂量的3,5-二苯基哒嗪-4-甲酸甲酯(10mg/kg)(其由于溶解度限制而以1ml/kg的体积施用)外,以0.5ml/kg的体积施用化合物。
2217、组织准备和分析
2218、在示踪剂注射后20分钟通过颈椎脱位处死大鼠。将完整的脑快速取出,并用无菌水轻轻漂洗。将主干血液收集进edta包被的微量离心管中,并储存在湿冰上直到研究结束。将海马和小脑剖离并在1.5ml微量离心管中保存,并置于湿冰上直到组织提取。在药物原初(drug naive)大鼠中,收集6个皮质脑组织样品用于产生空白和标准曲线样品。
2219、将含有0.1%甲酸的乙腈以4倍于组织样品重量的体积加入每个样品中。就标准曲线(0.1-30ng/g)样品而言,将计算的标准品体积减去乙腈体积。将样品匀浆化(fastprep-24,lysing matrix d;5.5m/s,进行60秒;或使用声波探头测探仪的7-8瓦功率;fisherscientific),并在14,000rpm离心16-分钟。用300μl无菌水(ph 6.5)稀释(100μl)上清液溶液。然后将该溶液彻底混合,并通过lc/ms/ms分析ro 15-4513(示踪剂)和3,5-二苯基哒嗪-4-甲酸甲酯。
2220、就血浆暴露而言,将血液样品在14000rpm离心16分钟。离心后,将来自每个样品的50ul上清液(血浆)加入200μl乙腈+0.1%甲酸中。就标准曲线(1-1000ng/ml)样品而言,将计算的标准品体积减去乙腈体积。将样品在超声水浴中声处理5分钟,然后在16000rpm离心30分钟。从每个样品小瓶中取出100ul上清液并放入新玻璃自动采样小瓶中,随后加入300μl无菌水(ph 6.5)。然后将该溶液彻底混合,并通过lc/ms/ms分析3,5-二苯基哒嗪-4-甲酸甲酯。
2221、通过对比海马(高gabaaα5受体密度区域)中的占有率和小脑(具有低gabaaα5受体密度的区域)中的占有率的比例方法确定受体占有率,并另外通过高剂量的gabaaα5负变构调节剂l-655,708(10mg/kg,静脉内)定义完全占有率。
2222、施用媒介物,随后施用示踪剂(1μg/kg的ro 15-4513,静脉内),导致与小脑(0.36±0.02ng/g)相比在海马(1.93±0.05ng/g)中高>5倍水平的ro 15-4513。3,5-二苯基哒嗪-4-甲酸甲酯(0.01-10mg/kg,静脉内)以剂量依赖性的方式减少在海马中的ro 15-4513结合,但是对于10mg/kg,静脉内的剂量没有影响ro 15-4513的小脑水平(图2),显示>90%的占有率(图3)。两种计算ro的方法产生了非常类似的结果,基于比例方法或使用l-755,608以定义占有率,3,5-二苯基哒嗪-4-甲酸甲酯的ed50值为1.8mg/kg或1.1mg/kg。
2223、在0.01mg/kg(静脉内)3,5-二苯基哒嗪-4-甲酸甲酯暴露在血浆和海马中低于定量限(bql),但在0.1mg/kg(静脉内)在海马中以低水平检测到(参见表3)。随着从0.1mg/kg至1mg/kg(静脉内)的10倍剂量增加,海马暴露是线性的,导致暴露的12倍增加。使剂量从1mg/kg增加至10mg/kg(静脉内),暴露仅增加~5倍。随着剂量从1mg/kg增加至10mg/kg(静脉内)时,血浆暴露增加12倍。
2224、表3:使用3,5-二苯基哒嗪-4-甲酸甲酯(0.01-10mg/kg,静脉内)的%gabaaα5受体占有率。在年轻long evans大鼠中治疗组的3,5-二苯基哒嗪-4-甲酸甲酯的海马和血浆暴露
2225、
2226、在老龄long-evans大鼠中进行另外的研究以确定在认知研究中行为相关剂量的暴露。还确定在年轻long-evans大鼠中的暴露以与在年轻long-evans大鼠中进行的受体占有率研究关联。在年轻和老龄long-evans大鼠中的暴露是相对类似的(表4,图4)。使剂量从1mg/kg至3mg/kg(静脉内)增加3倍,导致在年轻和老龄大鼠中在海马和血浆中的暴露大于剂量比例的增加,增加范围从4.5倍至6.6倍。
2227、表4:治疗组的3,5-二苯基哒嗪-4-甲酸甲酯在年轻long evans大鼠中的海马和血浆暴露
2228、
2229、在ro研究中,180ng/g在海马中的暴露(1mg/kg,静脉内)代表32-39%受体占有率,取决于用于确定ro的方法。该暴露与使用3mg/kg(静脉内)在老龄大鼠中观察到的结果相当,从而提示30-40%ro是在该模型中的认知效力所需要的。
2230、这些研究证实,3,5-二苯基哒嗪-4-甲酸甲酯产生了gabaaα5受体占有率的剂量依赖性增加。3,5-二苯基哒嗪-4-甲酸甲酯也表现出良好的脑暴露,脑/血浆比例>1。所述研究进一步证实,3,5-二苯基哒嗪-4-甲酸甲酯通过在gabaaα5亚型受体处的正变构调节产生它的认知增强作用。
2231、实施例108:3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯在老龄受损(ai)大鼠中的作用
2232、3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯(与achermann等人.bioorg.med.chem.lett.,19:5746-5752(2009)中的化合物编号49对应)是选择性的含有α5的gabaa r激动剂。
2233、使用媒介物对照品(25%环糊精,试验3次:在升/降系列的开始,中间和结束)和6种不同剂量水平(0.1mg/kg,0.3mg/kg,1mg/kg,3mg/kg,10mg/kg和30mg/kg,每种剂量试验2次)的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯,在与实施例107(a)中所述任务基本上类似的辐射臂迷宫(ram)行为任务中评估3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯对老龄受损(ai)大鼠的体内空间记忆保留的作用。使用相同的媒介物对照品和3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯剂量重复相同实验,其中将媒介物对照品试验5次,将3mg/kg剂量的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯试验4次,将其它剂量的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯试验2次。
2234、使用参数统计(配对t检验)对比在不同剂量的3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯和媒介物对照品的背景下ai大鼠在4小时延迟形式的ram任务中的保留试验性能(参见图5)。相对于媒介物对照品治疗,3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯在3mg/kg(t(7)=4.13,p=0.004,或t(7)=3.08,p=0.018)和在10mg/kg(t(7)=2.82,p=0.026)显著地改善记忆性能。
2235、还按照与在实施例107(b)中描述的程序(参见上面)基本上类似的程序研究了3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯对含有α5的gabaa受体占有率的影响。该研究证实,3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯(0.01-10mg/kg,静脉内)减少了海马中的ro 15-4513结合,对于10mg/kg(静脉内)的剂量没有影响ro 15-4513的小脑水平(图6),从而证实了>90%占有率(图7)。
2236、实施例109:使用莫里斯水迷宫行为任务6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮在老龄受损大鼠中的作用
2237、6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮(与chambers等人.j.med.chem.46:2227-2240(2003)中的化合物44对应)是选择性的含有α5的gabaa r激动剂。
2238、在莫里斯水迷宫行为任务中评估了6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮对老龄受损(ai)大鼠的体内空间记忆保留的作用。水迷宫是被一组相对于迷宫的新图案围绕的池。水迷宫的训练方案可以是基于已经被证实为海马依赖性的改进的水迷宫任务(de hoz等人,eur.j.neurosci.,22:745-54,2005;steele和morris,hippocampus 9:118-36,1999)。
2239、给认知受损的老龄大鼠在单侧用插管植入侧脑室中。脑功能区定位坐标是在前囟点后1.0mm,在中线侧面1.5mm和在头盖骨表面腹侧3.5mm。恢复约1周后,将大鼠在水迷宫中预训练2天(每天6次试验)以定位隐藏在池表面下面的浸没逃逸平台,其中逃逸平台位置每天改变。在预先训练过程中不施用脑室内(icv)输注。
2240、在预先训练后,在水迷宫训练和试验之前40分钟,大鼠接受在5μldmso中的100μg6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮(n=6)或媒介物dmso(n=5)的icv输注。训练由每天8次试验,持续2天组成,其中隐藏的逃逸平台保持在相同位置。给大鼠60秒以定位平台,其中有60秒试验间的间隔期。在训练结束后24h给大鼠施用探针试验(120秒),其中取出逃逸平台。在训练过程中,存在4个块,其中每个块具有4次训练试验。
2241、用媒介物和6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮治疗的大鼠在训练开始时几乎同时发现逃逸平台(块1)。在该块的训练中,用媒介物和6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮治疗的大鼠均花费约24秒找到逃逸平台。然而,用6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮治疗的大鼠能够在训练结束时(块4)比用单独媒介物治疗的那些大鼠更熟练地(即更快)找到平台。在块4中,用6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮治疗的大鼠花费约9.6秒找到逃逸平台,而用媒介物治疗的大鼠花费约19.69秒。这些结果提示,6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮改善了大鼠的水迷宫任务的学习(参见图8(a))。
2242、在训练后24h的试验测定中,取出逃逸平台。使用大鼠的探查/游泳模式来测量大鼠是否记住了在试验前训练中逃逸平台所定位的位置,以便试验大鼠的长期记忆力。在该试验中,"目标环形区域"是在试验前训练中平台所在区域周围的逃逸平台的大小的1.5倍的指定区域。"相对的环形区域"是与目标环形区域相同大小的对照区域,其位于池中目标环形区域的对侧。如果大鼠具有良好的长期记忆力,则将倾向于探查在试验前训练中平台所在位置周围的区域(即"目标"环形区域;而不是"相对的"环形区域)。"在环形区域中的时间"是以秒计的大鼠花费在目标或相对的环形区域面积中的时间的量。在环形区域中的"交叉通过次数(#)"是大鼠游泳穿过目标或相对的环形区域面积的次数。
2243、接受媒介物的大鼠花费相同的时间在目标环形区域和相对的环形区域中,从而表明这些大鼠似乎没有记住在试验前训练中平台所在的位置。相反,与它们花费在"相对的环形区域"中的时间或它们在"相对的环形区域"中交叉穿过的次数相比,用6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮治疗的大鼠在目标环形区域中花费显著更多的时间,并且更经常地交叉穿过"目标环形区域"。这些结果提示,6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮改善了大鼠在水迷宫任务中的长期记忆(参见,图8(b)和8(c))。
2244、本发明的化合物表现出对gabaaα5受体的正变构调节作用(参见,例如,实施例106)。这些化合物将增加gaba在gabaaα5受体处的作用。因此,本发明的化合物将在老龄受损动物(诸如大鼠)中产生认知增强作用,类似于其它gabaaα5受体选择性激动剂所产生的作用,诸如3,5-二苯基哒嗪-4-甲酸甲酯,3-甲氧基-7-甲基-9h-苯并[f]咪唑并[1,5-a][1,2,4]三唑并[4,3-d][1,4]二氮杂环庚三烯-10-甲酸乙酯和6,6二甲基-3-(3-羟基丙基)硫-1-(噻唑-2-基)-6,7-二氢-2-苯并噻吩-4(5h)-酮(参见,例如,实施例28-30)。
- 还没有人留言评论。精彩留言会获得点赞!