一种大麻二酚衍生物及其制备方法与应用
本发明涉及医药,尤其涉及一种大麻二酚衍生物及其制备方法与应用。
背景技术:
1、疼痛是一种复杂的生理心理活动,为临床常见症状之一。根据全球疼痛指数,身体疼痛普遍存在,但目前的疼痛治疗提供的缓解持续时间和缓解程度仍然不足。目前已有的治疗疼痛的药物包括阿片类药物、非类固醇消炎药、选择性cox2抑制剂(coxibs)、抗抑郁药、抗惊厥药和局部麻醉剂等,然而,这些药物有很大的局限性,包括便秘、耐受性和依赖性。因此,探索有效的新型镇痛机制及药物来治疗严重疼痛仍然非常重要。
2、辣椒素受体trpv1(transient receptor potential vanilloid 1)是一种配体门控的非选择性阳离子通道,是一种多模式伤害传感器,主要在中枢和外周的中、小型感觉神经元末端表达,在小脑、下丘脑、大脑皮质、嗅球、中脑、海马等不同的脑区域中也都有发现。辣椒素受体trpv1同其他trp离子通道亚家族内其他成员一样可以被质子、热、内源性配体激活,不同的是只有trpv1亚型能够被香草酸类化合物如辣椒素(capsaicin,cap)、树脂毒素(resiniferatoxin,rtx)激活。trpv1的激活会触发钙离子和钠离子的流入,从而引发一系列与疼痛传递相关的事件,包括膜去极化、神经元放电和疼痛递质的释放。由辣椒素受体激动剂产生的镇痛活性,由于不作用于阿片受体,不产生成瘾性而受到广泛关注。但是目前已知的香草酸类激动剂通常存在较为严重的刺激性副作用。
3、大麻二酚cbd是大麻中含量最丰富的非精神活性大麻素。cbd对大麻素受体的亲和力很低,但可能具有显著的cb1、cb2非依赖的作用机制,并在四氢大麻酚thc存在时具有拮抗cb1受体的作用。据报道,cbd是trpv1和5-ht1a受体的激动剂,并能够增强腺苷受体信号,可产生广泛的药理活性,包括镇痛、抗惊厥、抗炎、抗氧化和抗精神病作用。目前尚无cbd针对辣椒素受体trpv1的结构改造工作报道。
技术实现思路
1、本发明的目的为提供一种大麻二酚衍生物及其制备方法与应用,以解决现有技术针对辣椒素受体trpv1的大麻二酚衍生物的研究存在空白的问题。
2、为了达到上述目的,本发明采用如下技术方案:
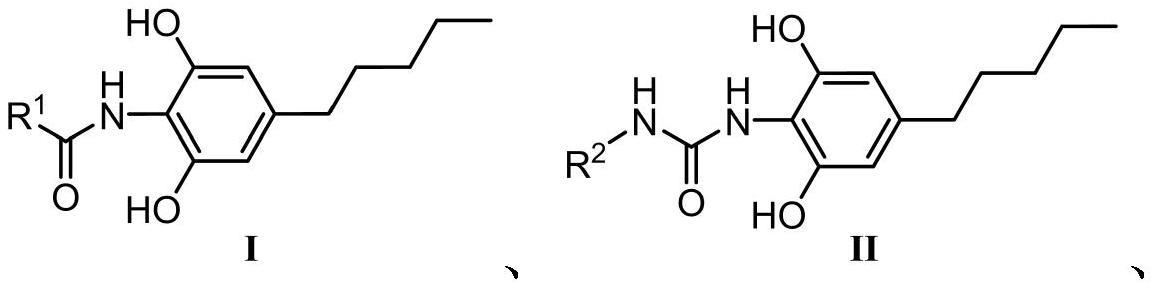
3、本发明提供了一种大麻二酚衍生物,所述大麻二酚衍生物为化合物或化合物的药学上可接受的盐;所述化合物的结构式为式ⅰ、式ⅱ、式ⅲ或式ⅳ;
4、所述式ⅰ为:所述式ⅱ为:所述式ⅲ为:所述式ⅳ为:
5、其中,r1、r2、r3、r4独立地优选为烃基、芳基或樟脑基;进一步地,所述r1、r2、r3独立地优选为取代芳基、喹啉、吡啶、咪唑基、环己基、金刚烷基、二苯甲酮基或樟脑基,进一步优选为4-喹啉基、4-苯甲酰基苯基、2-(3-溴吡啶基)、环己基、4-溴苯基、3-(1-(4-甲基四氢吡喃)吡唑基)、5-(2h-吡喃-2-酮基)、1-金刚烷基、2-(3-溴-5-氟吡啶基)、1-(4-溴苯基)环丁烷基、α-环己基-4-甲基苯基亚甲基、4-三氟甲氧基苯基、樟脑基、5-二甲氨基萘基、3,5-二氟苯基、联苯基、3,4-二氟苯基、3-三氟甲基-4-氟苯基、3-氟苯基、3-三氟甲基苯基或2,5-二氟苯基;r4优选为c1或c2邻苯二酚及其甲基保护化合物中的一种,所述c1或c2邻苯二酚及其甲基保护化合物中,c1或c2指的是邻苯二酚基团与酰胺上氮原子之间的碳原子数量;甲基保护的化合物指的是邻苯二酚基团上的羟基被甲基保护。
6、在本发明中,所述大麻二酚衍生物优选为选自如下所示结构的化合物或所述化合物的药学上可接受的盐:
7、
8、
9、在本发明中,所述药学上可接受是指适用于人而无过度不良副反应(如毒性、刺激和变态反应),即有合理的效益和风险比的物质。
10、在本发明中,所述药学上可接受的盐为所述化合物与药学上可接受的无机酸或有机酸所形成的盐,所述的无机酸可选自盐酸、氢溴酸、氢碘酸、磷酸、硝酸或硫酸;所述的有机酸可选自甲酸、乙酸、三氟乙酸、三氯乙酸、丙酸、丁二酸、甲磺酸、1,5-萘二磺酸、亚细亚酸、草酸、酒石酸、乳酸、水杨酸、苯甲酸、戊酸、二乙基乙酸、丙二酸、琥珀酸、富马酸、庚二酸、己二酸、马来酸、苹果酸、氨基磺酸、苯丙酸、葡糖酸、抗坏血酸、烟酸、异烟酸、苯磺酸、甲磺酸、对甲苯磺酸、柠檬酸、肉桂酸、丙酮酸、edta或氨基酸。
11、本发明还提供了所述1-a~10-a的制备方法,包括如下步骤:
12、(1)将化合物1与溴代试剂反应4~6h得到化合物2;(2)将化合物2与硼酸试剂反应得到化合物3;(3)将化合物3、r1-cooh与缩合试剂反应,后进行甲基脱除9~11h得到1-a~10-a。
13、在本发明所述步骤(1)中,所述化合物1的结构式优选为:所述溴代试剂优选为液溴、溴水、n-溴代丁二酰亚胺和三溴化吡啶鎓中的一种或多种,进一步优选为液溴和/或溴水;所述化合物1与溴代试剂的质量体积比优选为5~5.5g:2~2.3ml,进一步优选为5.1~5.4g:2.1~2.2ml;
14、本发明中,将化合物1、溴代试剂与反应溶剂混合后再进行反应;所述混合的具体步骤为:将化合物1与反应溶剂混合,后滴加溴代试剂;所述混合的温度优选为-1~1℃,进一步优选为0℃;所述反应溶剂优选为四氢呋喃、二氯甲烷、甲苯和苯中的一种或多种,进一步优选为四氢呋喃和/或二氯甲烷;所述化合物1与反应溶剂的质量体积比优选为5~5.5g:300~350ml,进一步优选为5.1~5.4g:310~340ml;所述溴代试剂的滴加速度优选为0.5~2ml/min,进一步优选为1ml/min。
15、在本发明所述步骤(2)中,将化合物2与硼酸试剂回流反应过夜得到化合物3;
16、所述化合物2的结构式优选为:所述硼酸试剂优选为戊基硼酸和/或戊基硼酸酯;所述化合物2与硼酸试剂的质量比优选为1.2~1.8:1.1~1.2,进一步优选为1.3~1.5:1.15;
17、所述化合物2与硼酸试剂在碱性试剂、催化剂和反应溶剂的条件下反应;所述碱性试剂优选为碳酸铯、碳酸钾、醋酸钾、磷酸钾、碳酸钠和醋酸钠中的一种或多种,所述催化剂优选为氯化钯、醋酸钯、四(三苯基膦)钯、双二苯基膦二茂铁二氯化钯和双二亚苄基丙酮钯的一种或多种,所述反应溶剂优选为四氢呋喃、二氧六环、甲苯、苯和二甲亚砜中的一种或多种;所述化合物2与反应溶剂的质量体积比优选为1~2g:50~100ml,进一步优选为1.2~1.5g:60~70ml;所述化合物2、碱性试剂与催化剂的质量比优选为1~2:4~5:0.2~0.5,进一步优选为1.2~1.5:4.1~4.3:0.3~0.45。
18、在本发明所述步骤(3)中,所述化合物3的结构式优选为:所述r1-cooh中的r1选自
19、所述缩合试剂优选为二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、o-(7-氮苯并三氮唑)-n,n,n,n-四甲基脲六氟磷酸酯和卡特缩合剂中的一种或多种;
20、所述化合物3与r1-cooh的摩尔比优选为0.4~0.7:0.6,进一步优选为0.5~0.6:0.6;所述化合物3与缩合试剂的质量比优选为10~12:28~29,进一步优选为11:28.1~28.5。
21、本发明中,将化合物3、r1-cooh与缩合试剂反应的具体步骤为:将r1-cooh的溶液、缩合试剂与碱性试剂反应20~40min后加入化合物3反应过夜;所述碱性试剂优选为叔丁醇钾、三乙胺、n,n-二异丙基乙胺、吡啶和1,8-二氮杂二环十一碳-7-烯中的一种或多种;所述r1-cooh的溶液为r1-cooh的n,n-二甲基甲酰胺溶液,r1-cooh与n,n-二甲基甲酰胺的摩尔体积比优选为0.5~0.8mmol:5~8ml,进一步优选为0.6~0.7mmol:6~7ml。
22、本发明所述1-a~10-a的制备路线为:
23、
24、本发明还提供了所述1-b、2-b、4-b或8-b的制备方法,包括如下步骤:将化合物3与反应试剂反应3~12h,后进行甲基脱除9~11h得到1-b、2-b、4-b或8-b;所述化合物3的制备方法同1-a~10-a中化合物3;
25、在本发明中,所述化合物3的结构式优选为:所述反应试剂优选为r2-nhcooph、r2-n=c=o或r2-nh2;当反应试剂为r2-nhcooph或r2-n=c=o时,反应在碱性试剂条件下进行,碱性试剂优选为叔丁醇钾、三乙胺、n,n-二异丙基乙胺、吡啶、2,6-二甲基吡啶和1,8-二氮杂二环十一碳-7-烯中的一种或多种;当反应试剂为r2-nh2时,反应在缩合试剂条件下进行,缩合试剂优选为二环己基碳二亚胺、n,n’-二琥珀酰亚胺基碳酸酯、1,1’-羰基二咪唑和2-氯-4,6-二甲氧基三氮唑中的一种或多种;所述甲基脱除所用试剂优选为三氯化铝、三溴化硼、三氯化硼和甲基碘化镁中的一种或多种;所述r2-nhcooph、r2-n=c=o或r2-nh2中的r2独立的选自
26、所述化合物3与反应试剂的摩尔比优选为0.1~1.5:0.3~1.1,进一步优选为0.5~1:0.5~1。
27、本发明所述1-b、2-b、4-b或8-b的制备路线为:
28、
29、本发明还提供了所述9-sn~20-sn的制备方法,包括如下步骤:将化合物3、r3-so2cl与碱性试剂反应7~9h,后进行甲基脱除9~11h得到9-sn~20-sn;所述化合物3的制备方法同1-a~10-a中化合物3;
30、在本发明中,所述化合物3的结构式优选为:所述r3-so2cl中的r3选自
31、所述碱性试剂优选为叔丁醇钾、三乙胺、n,n-二异丙基乙胺、吡啶和1,8-二氮杂二环十一碳-7-烯中的一种或多种;
32、所述化合物3、r3-so2cl与碱性试剂的摩尔比优选为0.2~0.3:0.25~0.3:0.6~0.7,进一步优选为0.21~0.28:0.26~0.28:0.61~0.68。
33、本发明所述9-sn~20-sn的制备路线为:
34、
35、本发明还提供了所述13-sn-1~13-sn-4的制备方法,包括如下步骤:
36、将化合物2、丙烯酸乙酯与催化剂在70~90℃下进行20~24h偶联反应得到化合物10;将化合物10在氢气氛围中进行3~5h氢化反应得到化合物11;将化合物11、r3-so2cl与碱性试剂进行7~9h缩合反应得到化合物12;将化合物12和水解试剂反应7~9h得到化合物13;将化合物12在0~5℃下进行9~11h甲基脱除得到13-sn-1;将化合物13在0~5℃下进行9~11h甲基脱除得到13-sn-2;将化合物13与缩合试剂进行11~13h缩合反应后在0~5℃下进行9~11h甲基脱除得到13-sn-3或13-sn-4;所述化合物2的制备方法同1-a~10-a中化合物2;
37、在本发明中,所述化合物2的结构式优选为:所述催化剂优选为氯化钯、醋酸钯、四(三苯基膦)钯、双二苯基膦二茂铁二氯化钯、双二亚苄基丙酮钯和三(二亚苄基丙酮)二钯的一种或多种;所述偶联反应在碱性试剂下进行,碱性试剂优选为叔丁醇钾、三乙胺、n,n-二异丙基乙胺、吡啶、2,6-二甲基吡啶和1,8-二氮杂二环十一碳-7-烯中的一种或多种;
38、所述化合物2与丙烯酸乙酯的质量体积比优选为1g:0.5~1ml,进一步优选为1g:0.6~0.8ml;所述化合物2与催化剂的摩尔比优选为4~4.5:0.1~0.5,进一步优选为4.1~4.3:0.2~0.4;所述化合物2与碱性试剂的质量体积比优选为1g:2~3ml,进一步优选为1g:2.2~2.7ml。
39、在本发明中,所述化合物10的结构式优选为所述氢化反应在催化剂下进行,催化剂优选为pd/c催化剂;
40、所述化合物10与催化剂的质量比优选为1~1.1:0.5,进一步优选为1.02~1.06:0.5。
41、在本发明中,所述化合物11的结构式优选为所述r3-so2cl中的r3选自所述碱性试剂优选为叔丁醇钾、三乙胺、n,n-二异丙基乙胺、吡啶和1,8-二氮杂二环十一碳-7-烯中的一种或多种;
42、所述化合物11、r3-so2cl与碱性试剂的摩尔比优选为1.1~1.2:1.7~1.8:3.5~4,进一步优选为1.15~1.18:1.72~1.78:3.6~3.9。
43、在本发明中,所述化合物12的结构式优选为所述水解试剂优选为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钠、碳酸钾和叔丁醇钾中的一种或多种;所述化合物13的结构式优选为
44、所述化合物12和水解试剂的摩尔比优选为1.1~1.2:11~12,进一步优选为1.13~1.18:11.2~11.5。
45、在本发明中,所述化合物12、化合物13和缩合反应所得产物进行甲基脱除所用试剂独立的优选为三氯化铝、三溴化硼、三氯化硼和甲基碘化镁中的一种或多种,进一步优选为三氯化铝、三溴化硼或三氯化硼;
46、在本发明中,所述缩合试剂优选为二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基苯并三唑、o-(7-氮苯并三氮唑)-n,n,n,n-四甲基脲六氟磷酸酯和卡特缩合剂中的一种或多种;所述化合物13与缩合试剂的摩尔比优选为1:2~2.5,进一步优选为1:2.2~2.4。
47、本发明还提供了所述13-sn-5~13-sn-7的制备方法,包括如下步骤:将13-sn-2、氨基化合物与缩合试剂反应11~13h得到13-sn-5~13-sn-7;所述13-sn-2为上述制备方法得到的13-sn-2。
48、在本发明中,所述氨基化合物优选为盐酸多巴胺、3,4-二甲氧基苄胺、4-羟基苄胺或5-(氨基甲基)-2-甲氧基苯酚盐酸盐;所述缩合试剂优选为二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基苯并三唑、o-(7-氮苯并三氮唑)-n,n,n,n-四甲基脲六氟磷酸酯和卡特缩合剂中的一种或多种;
49、所述13-sn-2、氨基化合物与缩合试剂的摩尔比优选为1.5~2.5:1.5~3:3~6,进一步优选为1.8~2:2~2.5:4~5。
50、本发明所述13-sn-1~13-sn-4的制备路线为:
51、
52、在本发明上述步骤中,所述甲基脱除的具体步骤为:将反应所得产物与反应试剂混合后滴加甲基脱除所用试剂;所述反应试剂独立地优选为四氢呋喃、二氯甲烷、甲苯和苯中的一种或多种;所述甲基脱除所用试剂独立地优选为三氯化铝、三溴化硼、三氯化硼和甲基碘化镁中的一种或多种,进一步优选为三溴化硼和/或三氯化硼;所述反应所得产物与反应试剂混合后所得混合液的浓度独立地优选为0.01~0.2mm,进一步优选为0.15mm;所述甲基脱除所用试剂的用量独立地优选为3~8当量,进一步优选为5当量;所述甲基脱除所用试剂的滴加速度独立地优选为1~5ml/min,进一步优选为3ml/min;所述滴加的温度独立地优选为-80~-75℃,进一步优选为-78~-76℃。
53、在本发明所述1-a~10-a的制备步骤中,步骤(1)和(2)中,反应后将反应所得产物进行后处理;步骤(2)中的后处理具体为:将反应所得产物顺次进行抽滤、淋洗、减压浓缩和分离纯化,所述淋洗所用试剂优选为乙酸乙酯;步骤(3)中,甲基脱除前对反应所得产物进行后处理;甲基脱除后对所得产物进行后处理。
54、在本发明所述1-b、2-b、4-b或8-b的制备步骤中,甲基脱除前对反应所得产物进行后处理;甲基脱除后对所得产物进行后处理。
55、在本发明所述9-sn~20-sn的制备步骤中,偶联反应后对反应所得产物进行后处理,后处理的具体步骤为:将所得产物顺次进行抽滤、淋洗、洗涤、干燥、抽滤、减压浓缩和分离纯化,所述淋洗所用试剂优选为乙酸乙酯;氢化反应后对反应所得产物进行后处理,后处理的具体步骤为:将所得产物顺次进行抽滤、淋洗和减压浓缩;所述淋洗所用试剂优选为乙酸乙酯;缩合反应后对反应所得产物进行后处理;水解反应后对反应所得产物进行后处理。
56、在本发明所述13-sn-1~13-sn-4的制备步骤中,得到13-sn-1~13-sn-4前对甲基脱除所得产物进行后处理。
57、在本发明所述13-sn-5~13-sn-7的制备步骤中,反应后对反应所得产物进行后处理。
58、在本发明上述步骤中,除所述1-a~10-a的制备步骤(2)、所述1-b、2-b、4-b或8-b的制备步骤中的偶联反应以及氢化反应外,其余步骤中的后处理的具体步骤为:将所得产物顺次进行淬灭、洗涤、干燥、抽滤、减压浓缩和分离纯化。
59、在本发明中,淬灭所用试剂独立地优选为饱和nahco3溶液、水和乙酸乙酯的混合液或水;洗涤所用试剂优选为饱和nacl和/或水,洗涤的次数独立地优选为2~4次,进一步优选为3次;干燥优选为采用无水硫酸钠进行;所述分离纯化优选为硅胶柱色谱分离纯化;
60、在本发明中,所述淬灭或淋洗均是为了分离有机相和水相;淬灭或淋洗所得水相进行萃取得到有机相,与淬灭所得有机相合并进行洗涤;所述萃取所用试剂优选为二氯甲烷。
61、本发明还提供了所述大麻二酚衍生物或所述大麻二酚衍生物的制备方法制备得到的大麻二酚衍生物在制备辣椒素受体产生镇痛效应的药物中的应用。
62、在本发明中,所述药物的剂型不限,只要是能够使活性成分有效地到达体内的剂型即可,所述药物的剂型可选自片剂、胶囊剂、粉末、颗粒剂、糖浆、溶液、悬浮液、注射剂、酊剂、口服液、气雾剂、口含剂、冲剂、丸剂、散剂或纳米制剂。
63、在本发明中,所述药物中除了含有主要活性成分之外,还可含有少量的且不影响有效成分的次要成分和/或药学上可接受的载体;所述药物中可以含有甜味剂以改善口味、抗氧化剂以防止氧化,以及各种制剂所必要的辅料。
64、在本发明中,所述药物的活性成分的有效施用剂量可随所用的药物、给药的模式和待治疗的疾病的严重程度而变化。
65、经由上述的技术方案可知,与现有技术相比,本发明有益效果如下:
66、本发明所述的大麻二酚衍生物,对于辣椒素受体(trpv1)产生镇痛活性具有显著作用,可用于制备新型作用模式的镇痛药物,具有广泛应用前景。
- 还没有人留言评论。精彩留言会获得点赞!