一种交联聚维酮及其制备方法和应用与流程
本发明涉及交联聚维酮,尤其涉及一种交联聚维酮及其制备方法和应用。
背景技术:
1、交联聚维酮(pvpp)产品是一种超强吸水性树脂,具有很强的吸水、保水能力。其吸水能力可达到自身重量的数倍,甚至数十倍,并且基体与吸附水间能形成氢键,即使在一定压力下吸附水也不会流失,可回收重复循环使用。pvpp好的生理安全性、吸水性、水不溶性、络合性等优良特性。
2、交联聚维酮(pvpp)产品,通过独特的爆米花聚合工艺,形成多孔型、颗粒状的非离子型产品,具有较强的界面活性。爆米花聚合是一种特殊的聚合方式,其反应机理现在还不清楚,爆米花聚合得到的聚合物是高度交联的,但它的交联主要是由长分子链之间相互缠绕形成的。目前一般使用碱金属氢氧化物、纯化水、1-乙烯基-2-吡咯烷酮(nvp)通过爆米花聚合的方法来生产交联聚维酮,但是此方法由于过程中会产生副反应,杂质含量高,收率低。
3、在传统pvpp制备工艺中,pvpp的粒径可控范围较小,基本只能控制在小于100μm的范围内,这样极大的限制了pvpp在酿酒和饮料行业中作为澄清剂、稳定剂的应用效果,同时能够重复使用的次数较少,造成资源的浪费;此外,对于用于压片制备无尘交联聚维酮颗粒应用时,粒径太细,可压性不足,严重影响生产效率和设备使用寿命。
4、cn106883333a公开了一种大粒径交联聚维酮的制备方法,在单体nvp:水的质量比为9:1的条件下,加入3.0%碱金属氢氧化物,通入氮气保护,加热至55℃引发进行爆米花聚合,搅拌速度60转/分;在温度上升到70℃时,加入0.5%~5.0%的成型剂,同时降低搅拌速度至20转/分;颗粒成型后,经过洗涤干燥,最终得到粒径在140~200μm之间交联聚维酮;成型剂一般为叔丁基过氧化氢、过氧化苯甲酰、二叔丁基过氧化物、偶氮二异丁腈、过氧化苯甲酸叔丁酯中的至少一种。从其技术方案,可以看出,在聚合之后添加的叔丁基过氧化氢、过氧化苯甲酰、二叔丁基过氧化物、偶氮二异丁腈、过苯甲酸特丁酯都是常用的nvp引发剂,实际上是起到消残作用的助剂,将交联聚合阶段未反应的nvp单体引发反应生成pvp,实际是pvp起到的表面活性剂的作用,改变颗粒成型时的表面张力,来达到大粒径的pvpp制备的目的。暂且不论该技术方案能否达成所谓的大粒径的pvpp的制备,很显然其公开的方法是无法保证收率的,因为交联聚合阶段未反应的nvp单体引发反应生成pvp是水溶性的,在后续清洗处理阶段都会溶于水中而流失掉的。
5、cn111378060a公开了一种基于原位生成交联剂的爆米花聚合机理进行交联聚维酮的生产方法,属于药用辅料技术领域。包括以下步骤:1)聚合反应;2)浸泡工序;3)离心脱水;4)干燥,制得最终产品。但是,其公开的工艺需要经过浸泡工序处理,用到大量的冰乙酸、过氧乙酸,且工艺耗时长,仅浸泡工序就需要至少20-60h,且处理比较复杂;使用大量的酸浸泡清洗,产生大量的强酸强碱的工业污水,工艺高污染,高能耗,经济效益低。
6、综上所述,开发一种能克服上述缺陷的交联聚维酮及其制备方法是至关重要的。
技术实现思路
1、针对现有技术的不足,本发明的目的在于提供一种交联聚维酮及其制备方法和应用,所述交联聚维酮粒径大,而且吸水快速、水合性能好,所述制备方法的无需耗用大量的酸进行酸洗处理,产废少,工艺能耗低,成本低,污染低,绿色环保,形成的交联聚维酮杂质含量低,收率高。
2、为达此目的,本发明采用以下技术方案:
3、第一方面,本发明提供一种交联聚维酮,所述交联聚维酮的制备原料包括1-乙烯基-2-吡咯烷酮、氨水、尿素、碱金属氢氧化物和第一溶剂。
4、本发明中,尿素作为催化剂和改性剂,氨水作为结构改性剂和晶核保护剂,形成的交联聚维酮粒径大,具有优异的吸水能力、崩解性能和水合性能,综合性能优异。
5、优选地,所述1-乙烯基-2-吡咯烷酮、氨水、尿素、碱金属氢氧化物和第一溶剂的质量比为100:(0.1~5.0):(1.0~5.0):(0.5~1.25):(30~40);其中,0.1-5.0可以为0.2、0.5、0.8、1.0、2.0、3.0、4.0、5.0等;1.0-5.0可以为1.0、2.0、3.0、4.0、5.0等;0.5-1.25可以为0.6、0.8、1、1.2等;30-40可以为32、34、36、38等。
6、优选地,所述氨水的质量浓度为20.0%-30.0%,例如21%、22%、23%、24%、25%、26%、27%、28%、29%等。
7、优选地,所述碱金属氢氧化物包括氢氧化锂、氢氧化钠或氢氧化钾中的任意一种或至少两种的组合,其中典型但非限制性的组合包括:氢氧化锂和氢氧化钠的组合,氢氧化钠和氢氧化钾的组合,氢氧化锂、氢氧化钠和氢氧化钾的组合等。
8、优选地,所述第一溶剂包括水。
9、优选地,所述交联聚维酮中,粒径为100-500μm(例如150μm、200μm、250μm、300μm、350μm、400μm、450μm等)的交联聚维酮占比≥90%,例如92%、94%、96%、98%等,进一步优选90.03%~93.26%。
10、第二方面,本发明提供一种第一方面所述的交联聚维酮的制备方法,所述制备方法包括如下步骤:
11、将1-乙烯基-2-吡咯烷酮、氨水、尿素、碱金属氢氧化物和第一溶剂混合,再进行聚合反应、打浆、清洗和消残处理,得到所述交联聚维酮。
12、本发明中,尿素在制备中起到催化剂和改性剂的双重作用效果:第一方面,尿素在碱性聚合液环境中高温加热分解产生氨,在反应中能够提升nvp的乙烯基基团的聚合活性,加快交联剂的生成,加快链引发交联聚合反应进行,提升反应速率;第二方面,尿素分子为碳酰胺结构小分子,在聚合反应过程中,改性链终止阶段的反应,形成rh2-o-c(nh2)2的封端结构,可以改善其吸水速率及吸附性能。所述消残处理的作用是分解或反应掉聚合物体系中的残留单体杂质,同时改善产品的酸碱度;氨水在制备中发挥的作用:主要是起到阻止或减少聚合高温阶段尿素水解反应的进行,间接起到结构改性剂和晶核保护剂的作用;本发明制备工艺中加入了以单体量计的30~40%的水,相对于现有技术,水的加入量明显偏高,主要是为聚合反应得到的改性交联pvpp晶核团聚长大提供水系环境,提升产物交联pvpp颗粒粒径。本发明所述的制备方法无需耗用大量的酸进行酸洗处理,产废少,工艺能耗低,成本低,污染低,绿色环保,形成的交联聚维酮杂质含量低。
13、优选地,所述聚合反应包括第一次升温反应、第二次升温反应和降温反应。
14、优选地,所述第一次升温反应至35-50℃,例如36℃、38℃、40℃、42℃、44℃、46℃、48℃等。
15、优选地,所述第一次升温后,除氧。
16、优选地,所述除氧的方式包括通惰性气体和抽真空,进行至少一次(例如两次、三次、四次等)。
17、优选地,最后一次通惰性气体和抽真空后,在体系中通惰性气体,保持体系压力为0-0.1mpa,例如0.02mpa、0.04mpa、0.06mpa、0.08mpa等。
18、示例性地,所述惰性气体包括氮气。
19、示例性地,所述除氧具体包括:启动搅拌,抽真空至-0.06~-0.1mpa(例如-0.07mpa、-0.08mpa、-0.09mpa等),保持5-15min(例如6min、7min、8min、9min、10min、11min、12min、13min、14min等);然后通氮气加压至0.2~0.3mpa(例如0.22mpa、0.24mpa、0.26mpa、0.28mpa等),保压5-15min(例如6min、7min、8min、9min、10min、11min、12min、13min、14min等);继续如此操作置换至少1次(例如两次、三次、四次等)后,通氮气加压至0-0.1mpa(例如0.02mpa、0.04mpa、0.06mpa、0.08mpa等)。
20、优选地,体系在800-1200转/min(例如900转/min、1000转/min、1100转/min等)下,进行第二次升温。
21、优选地,所述第二次升温至120-130℃,例如122℃、124℃、126℃、128℃等。
22、优选地,所述第二次升温后,保温反应。
23、优选地,所述保温反应的时间为30-90min,例如35min、40min、45min、50min、55min、60min、65min、70min、75min、80min、85min等。
24、优选地,所述保温反应后,降温至80-110℃(例如85℃、90℃、95℃、100℃、105℃等),降温中,温度再次出现上升,聚合反应开始。
25、本发明中,自然降温观察温度变化,自然降温过程中,温度再次出现上升,表明交联聚合反应开始。
26、优选地,所述聚合反应的温度为80-120℃,例如85℃、90℃、95℃、100℃、105℃、110℃、115℃等。
27、优选地,所述聚合反应的压力为0.1-0.6mpa,例如0.2mpa、0.3mpa、0.4mpa、0.5mpa等。
28、优选地,调整聚合反应后的物料温度至80-100℃(例如85℃、90℃、95℃等),向体系中加入第二溶剂混合,进行搅拌打浆。
29、优选地,以所述1-乙烯基-2-吡咯烷酮的总质量为100份计,所述打浆采用的第二溶剂的质量为300-500份,例如320份、340份、360份、380份、400份、420份、440份、460份、480份等。
30、优选地,所述第二溶剂包括水。
31、优选地,所述搅拌打浆的转速为50-150转/min,例如60转/min、80转/min、100转/min、120转/min、140转/min等。
32、优选地,所述搅拌打浆的时间为15-30min,例如16min、18min、20min、22min、24min、26min、28min等。
33、优选地,所述清洗在设置有400目~500目(例如420目、440目、460目、480目等)滤布的清洗过滤器中进行。
34、优选地,所述清洗包括将打浆后的物料抽真空脱除第一溶剂和第二溶剂。
35、优选地,所述清洗还包括将滤布截留的物料与第三溶剂混合,抽真空,所述操作至少进行一次(例如两次、三次、四次、五次等)。
36、优选地,所述滤布截留的物料与第三溶剂混合的方式包括搅拌。
37、优选地,以所述1-乙烯基-2-吡咯烷酮的总质量为100份计,所述混合采用的第三溶剂的添加量为300-500份,例如320份、340份、360份、380份、400份、420份、440份、460份、480份等。
38、优选地,所述第三溶剂包括温度为60-80℃的水,例如62℃、64℃、66℃、68℃、70℃、72℃、74℃、76℃、78℃等。
39、优选地,所述搅拌的转速为50-150转/min,例如60转/min、80转/min、100转/min、120转/min、140转/min等。
40、优选地,所述搅拌的时间为30-60min,例如35min、40min、45min、50min、55min等。
41、优选地,将所述清洗后的滤布截留的物料进行消残处理。
42、优选地,所述消残处理包括将所述清洗后的滤布截留的物料与第四溶剂混合,保温,加入消残助剂处理。
43、优选地,所述消残助剂包括过氧化氢、过氧乙酸、叔丁基过氧化氢或过氧乙酸叔丁酯中的任意一种或至少两种的组合,其中典型但非限制性的组合包括:过氧化氢和过氧乙酸的组合,过氧乙酸、叔丁基过氧化氢和过氧乙酸叔丁酯的组合,过氧化氢、过氧乙酸、叔丁基过氧化氢和过氧乙酸叔丁酯的组合等。
44、优选地,所述消残处理中采用的第四溶剂的添加量为300-500份,例如320份、340份、360份、380份、400份、420份、440份、460份、480份等。
45、优选地,所述第四溶剂包括水。
46、优选地,所述保温的温度为60-80℃,例如62℃、64℃、66℃、68℃、70℃、72℃、74℃、76℃、78℃等。
47、优选地,所述消残处理的时间为6-12h,例如7h、8h、9h、10h、11h等。
48、优选地,所述消残处理至体系1-乙烯基-2-吡咯烷酮的残留量≤10ppm,例如9ppm、8ppm、7ppm、6ppm、5ppm、4ppm等。
49、优选地,所述消残处理后,还包括脱除溶剂和干燥。
50、优选地,所述脱除溶剂的方式包括离心。
51、优选地,所述脱除溶剂至离心截留物料液中的水可溶物的质量含量≤1.0%,例如0.8%、0.6%、0.4%、0.2%等。
52、作为优选的技术方案,所述制备方法包括如下步骤:
53、(1)将碱金属氢氧化物与第一溶剂混合形成氢氧化物溶液;
54、(2)将1-乙烯基-2-吡咯烷酮、氨水、尿素、碱金属氢氧化物溶液混合,第一次升温至35-50℃,通惰性气体和抽真空,进行至少一次,在体系中通惰性气体,保持体系压力为0-0.1mpa,完成除氧;
55、(3)在800-1200转/min下进行升温,至120-130℃后,保温反应30-90min,降温至80-110℃,降温中,温度再次出现上升,聚合反应开始,控制温度至80-120℃,体系压力至0.1-0.6mpa,至聚合反应结束;
56、(4)调整体系温度至80-100℃时,将聚合反应后的物料与第二溶剂混合,在50-150转/min下进行搅拌打浆15-30min;
57、(5)将打浆后的物料转移至装有400目~500目的滤布的清洗过滤器中,抽真空脱除第一溶剂、第二溶剂和溶剂中可溶物;再将滤布截留的物料和第三溶剂在50-150转/min下搅拌混合30-60min,抽真空,所述操作至少进行一次,完成清洗;
58、(6)将滤布截留的物料与第四溶剂混合,在60-80℃下保温,消残处理6-12h,至体系1-乙烯基-2-吡咯烷酮的残留量≤10ppm,完成消残处理;
59、(7)将所述消残处理后的物料进行脱除溶剂和干燥,得到所述交联聚维酮。
60、第三方面,本发明提供一种无尘交联聚维酮,所述无尘交联聚维酮包括第一方面所述的交联聚维酮。
61、第四方面,本发明提供一种吸水树脂,所述吸水树脂包括第一方面所述的交联聚维酮。
62、相对于现有技术,本发明具有以下有益效果:
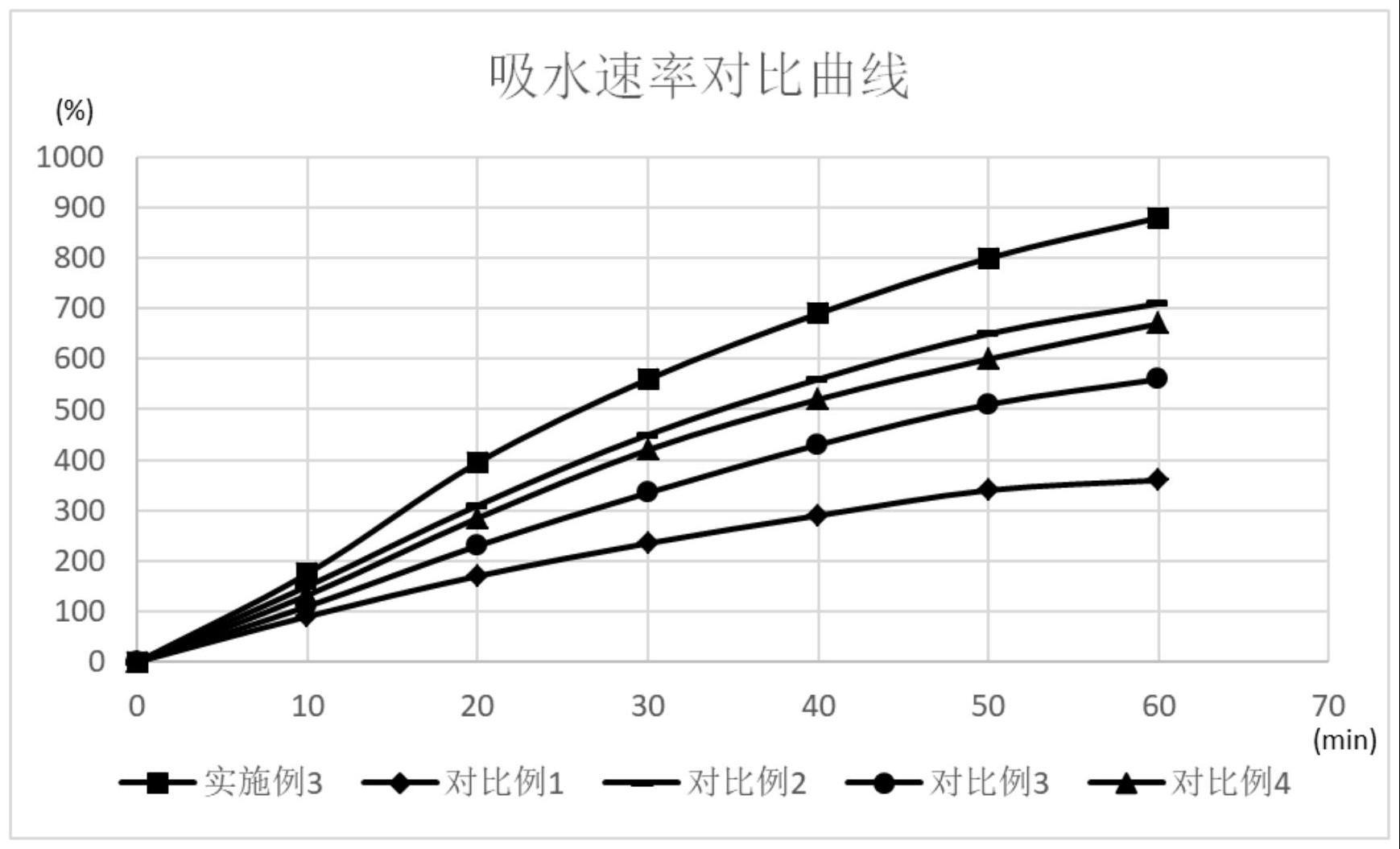
63、(1)本发明所述的交联聚维酮粒径在100-500μm之间占比在90.03%~93.26%;在吸水60min后,吸水率在810%以上,以所述交联聚维酮作为崩解剂,崩解时间在21.53min以内,水合能力在8.1以上
64、(2)本发明所述的交联聚维酮的理化指标能够满足cp、ep、usp、bp及jp的最高标准要求,杂质a(nvp)≤10ppm,水中可溶物≤1.5%,过氧化物≤400ppm。
65、(3)本发明所述的制备方法无需耗用大量的酸进行酸洗处理,产废少,工艺能耗低,成本低,污染低,绿色环保,形成的交联聚维酮杂质含量低,收率高,在95%以上。
- 还没有人留言评论。精彩留言会获得点赞!