一种新颖的氨基甲酸酯类化合物及其用途的制作方法
本发明属于药物化学领域,具体涉及一种氨基甲酸酯类化合物,以及所述化合物在制备抗血小板聚集药物中用途。
背景技术:
1、本领域公知,血小板聚集可以引发一系列心、脑血管及其他动脉循环障碍疾病,包括(1)急性冠状动脉综合征(acs),如不稳定型心绞痛(ua)、急性st段抬高性心肌梗死(stemi)和急性非st段抬高性心肌梗死(nstemi);(2)动脉粥样硬化疾病,如心肌梗死、缺血性卒中、外周动脉性疾病;(3)以及血栓性并发症等。针对目前的抗血小板凝集药物中,氯吡格雷因出血风险较小备受关注,但因其存在“氯吡格雷抵抗”现象大大限制了该药物的起效时间和应用场景,如急性血栓。虽然现有技术也尝试制备氯吡格雷的注射剂型,以期达到快速起效,克服急性治疗环境中起效慢等缺陷,如ascendia公司的asd-002纳米乳剂、cydex公司的mdco-157(环糊精包合)注射剂以及jina pharmaceuticals公司的jin-2013纳米脂质体注射剂;但至今因氯吡格雷的溶解度和水、光、热稳定性问题,均以失败告终。此外,以成都施贝康生物医药科技有限公司为首开发的氯吡格雷代谢中间产物2-氧代氯吡格雷,虽然克服了“氯吡格雷抵抗”不良反应,获得起效快速、生物利用度更高等优势;但在研究中发现,2-氧代氯吡格雷仍然存在水中不稳定性缺陷,对于开发成注射剂型的挑战和限制极大。因此,继而开发中间代谢物2-氧代氯吡格雷的衍生物引起了社会各界的广泛关注。
2、目前,以2-氧代氯吡格雷为基础的前药分子中仅维卡格雷进入临床,且是片剂形式。但在对比研究中发现,维卡格雷仍然存在诸多局限性,如溶解度和热稳定性等不理想,同样将很大程度限制其给药形式和临床应用场景。
3、综上,开发一种可体内持续长久释放出氯吡格雷活性代谢物成分(h4)且水溶性和稳定性较好的前药分子,同时解决氯吡格雷不可注射的特性,是目前临床亟待解决的难题。
技术实现思路
1、本发明的目的之一在于,提供一种水中溶解度优良的2-氧代氯吡格雷前药分子化合物。
2、本发明的目的之二在于,提供一种可用于预防和治疗因血小板聚集引起的心、脑及其他动脉循环障碍疾病的用途。
3、本发明的技术方案如下:
4、本发明提供一种式i所示的化合物,或其药学上可接受的盐、溶剂化物或氘代物:
5、。
6、进一步地,上述药学上可接受的盐包含富马酸盐、乙酸盐、抗坏血酸盐、苯甲酸盐、苯磺酸盐、柠檬酸盐、盐酸盐、氢溴酸盐、马来酸盐、甲磺酸盐、硫酸盐、硫酸氢盐、硝酸盐、草酸盐、磷酸盐或琥珀酸盐。
7、进一步地,上述药学上可接受的盐选自盐酸盐,其中盐酸与式i所示的化合物的摩尔比为1:1或2:1。
8、本发明提供一种含有上述任一所述的化合物,或其药学上可接受的盐、溶剂化物或氘代物的药物组合物,所述药物组合物还包含药学上可接受辅料。
9、本发明还提供上述化合物,或其药学上可接受的盐,溶剂化物或氘代物在制备预防和/或治疗因血小板聚集引起的心、脑及其他动脉循环障碍疾病中的药物中的用途。
10、进一步地,上述因血小板聚集引起的心、脑及其他动脉循环障碍疾病,包括但不限于急性冠脉动脉综合征、动脉粥样硬化疾病、或血栓性并发症。
11、进一步地,上述急性冠脉动脉综合征包括但不限于心绞痛或心肌梗死。
12、更进一步地,上述急性冠脉动脉综合征包括但不限于不稳定型心绞痛(ua)、急性st段抬高性心肌梗死(stemi)或急性非st段抬高性心肌梗死(nstemi)。
13、进一步地,上述动脉粥样硬化疾病包括但不限于心肌梗死、缺血性卒中或外周动脉性疾病。
14、更进一步地,上述缺血性卒中包括但不限于脑卒中。
15、进一步地,上述血栓性并发症包括但不限于肺梗死。
16、有益技术效果:
17、本发明的化合物具有更好的体外药效、更优的药代动力学特征。具体地,本发明的化合物具有较强的抗血小板聚集作用,起效快且体内药代的血药浓度和生物利用度高,半衰期较长,药效更优。此外,本发明的化合物体外溶解度好、稳定性高,溶血风险小、无心脏毒性且血管刺激风险小,安全性高,更有利于做成注射液,弥补临床不足。
18、实施方式
19、以下将结合实施例和试验例对本发明作进一步的详细描述,本发明的实施例和试验例仅用于说明本发明的技术方案,并非对本发明的限制,凡依照本发明公开的内容所作的任何本领域的等同置换,均属于本发明的保护范围。
20、本发明的化合物、其立体异构体或药学上可接受的盐均可选择实施例的合成路线进行制备,并根据取代基或成盐的需要,对反应原料和反应溶剂的常规条件加以调整,这些都是本领域的技术人员在本发明公开内容的基础上可以实现的。此外,本发明的柱层析在没有特别说明的情况下指硅胶柱层析,洗脱溶剂在没有特别说明的情况下可以结合反应溶剂与本领域技术人员的公知常识或者常用手段确定单一或者混合洗脱溶剂。
21、化合物的结构是核磁共振(1h nmr)或液质联用(lc-ms)来确定的。
22、液质联用仪(lc-ms)为安捷伦g6120b(与液相agilent 1260配用);核磁共振仪(1hnmr)为bruker avance-400或bruker avance-800,核磁共振(1h nmr)位移( δ)以百万分之一(ppm)的单位给出,测定溶剂为dmso-d6或cdcl3,内标为四甲基硅烷(tms),化学位移是以10-6(ppm)作为单位给出。
23、本发明的术语“室温”是指温度处于10~25℃之间。
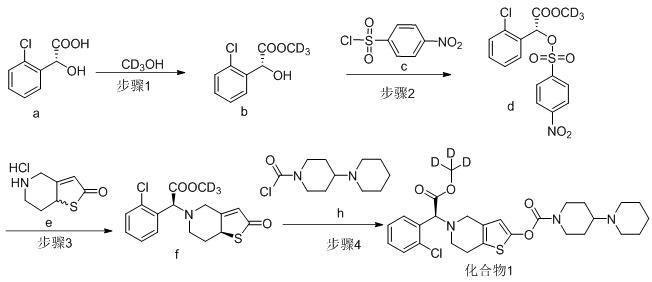
24、实施例1:(s)-5-(1-(2-氯苯基)-2-(甲氧基-d3)-2-氧乙基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基[1,4'-二哌啶]-1'-羧酸酯(化合物1)的制备
25、
26、步骤1:(r)-2-(2-氯苯基)-2-羟基乙酸甲-d3-酯(中间体b)的制备
27、室温下将4g (r)-邻氯扁桃酸投入50ml三口瓶中,加入12ml无水氘代甲醇搅拌溶解,后缓慢滴加0.17g浓硫酸。滴毕,以油浴加热将外温升至78℃回流2小时。tlc监控反应(展开剂:乙酸乙酯/石油醚=1/3,254nm紫外灯下显色)。以外温45℃减压浓缩除去甲醇,残留物补加12ml乙酸乙酯搅拌溶解。乙酸乙酯层以10mlⅹ2 水洗两遍,再分别以10 ml饱和碳酸氢钠,10ml饱和食盐水洗涤后,乙酸乙酯层以无水硫酸钠干燥,过滤。滤液以45℃外温减压浓缩除去乙酸乙酯,得微黄透明油状液体0.415g,即为中间体b。收率:95.1%。
28、esi-ms: m/z = 204.1(m+h) +。
29、1hnmr (400 mhz, cdcl3) δ:7.4 (m,1h),7.35 (m, 1h), 7.23 (m, 2h), 5.58(s, 1h) , 4.06 (brs, 1h)。
30、步骤2:(r)-2-(2-氯苯基)-2-(((4-硝基苯基)磺酰基)氧基)乙酸甲-d3-酯(中间体d)的制备
31、室温下将4.15g中间体i,15ml二氯甲烷投入30ml单口瓶中,冰浴下搅拌溶解,再加入2.51g三乙胺,降温并控制内温不高于5℃。以恒压滴液漏斗缓慢滴加以15ml二氯甲烷溶解的4.35g对硝基苯磺酰氯,1.5小时滴毕,滴毕后继续维持冰浴0.5小时。冰浴下以恒压滴液漏斗滴加4m 冰盐酸5ml淬灭反应,剧烈搅拌至体系呈明黄色后停止,分液。二氯甲烷层以10ml水,10ml×2饱和食盐水洗涤,并在35外温下减压浓缩除去二氯甲烷,得黄色膏状中间体d粗品。粗品投入50ml烧杯,在冰浴下以10ml冰甲醇打浆0.5小时,期间析出大量白色固体。过滤固体,得白色粉末状固体5.61g,即为中间体d,收率为70.8%。。
32、esi-ms: m/z = 389.1(m+h) +。
33、1hnmr (400 mhz, cdcl3) δ: 8.31 (d, j = 8.9 hz,2h),8.07 (d,j = 8.9 hz,2h), 7.31 (m, 4h), 6.39 (s, 1h)。
34、步骤3:(s)-2-(2-氯苯基)-2-((s)2-氧代-2,6,7,7a-四氢噻吩并[3,2-c]吡啶-5(4h)-基)乙酸甲-d3-酯(中间体f)的制备
35、反应:室温下将5.61g中间体b,3.19g 化合物e,5.39g无水碳酸钠以及100ml乙腈投入200ml三口瓶中,升温至38℃并在氮气的保护下剧烈搅拌反应5小时。反应液以硅藻土过滤,在外温45℃下减压浓缩除去乙腈。残留物以20ml二氯甲烷溶解,二氯甲烷层分别以10ml×2水,10ml饱和食盐水洗涤,分液。过滤,滤液减压浓缩,得部分析晶褐色膏状半固体4.18g,在60℃水浴下将4.18g sbk002粗品,10ml丙酮投入20ml烧杯中充分搅拌10分钟。基本溶清,趁热过滤,析晶,得类白色结晶性粉末2.95g,收率为60.0%。
36、esi-ms: m/z = 341.1(m+h) +。
37、步骤4:(s)-5-(1-(2-氯苯基)-2-(甲氧基-d3)-2-氧乙基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基[1,4'-二哌啶]-1'-羧酸酯(化合物1)的制备
38、50ml三口瓶中加入中间体f (1g,3mmol)和4-甲基哌嗪-1-甲酰氯盐酸盐(884mg,4.44mmol),dmf(20ml),搅拌降温至0℃,滴加入dbu(1.37g,9mmol)。滴加完毕,室温反应2h结束。加入ea,水洗二次,盐水洗涤一次。有机相用无水na2so4干燥且浓缩。浓缩物通过层析柱分离纯化,收集产物,浓缩得到1.2g化合物1,收率87.6%,纯度为98.34%。
39、esi-ms: m/z = 535.2(m+h) +。
40、1hnmr (400 mhz, dmso-d6) δ: 7.52~7.43 (m, 1h), 7.43~7.34 (m, 1h),7.32~ 7.21 (m, 2h), 6.29 (s, 1h), 4.75 (d, 1h), 3.90 (d, 2h), 3.69~ 3.59(m, 2h), 3.39 (m, 2h), 3.15~3.08 (m, 1h), 3.02 (t, 1h), 2.91 (t, 2h), 2.68(m, 2h), 2.39 (q, 1h), 2.31 (m, 2h), 1.74~1.37 (m, 10h)。
41、实施例2:(s)-5-(1-(2-氯苯基)-2-甲氧基-2-氧乙基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基4-甲基哌嗪-1-羧酸酯富马酸盐(化合物2)的制备
42、
43、25ml烧瓶中加入(s) -5-(1-(2-氯苯基)-2-(甲氧基-d3)-2-氧乙基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基-4-甲基哌嗪-1-羧酸酯(1 g,1.87mmol),加入ea(2.5ml)溶解,加入1.87mmol盐酸搅拌,大量固体析出。搅拌3h结束,过滤,烘干得到标题化合物671 mg。收率62.7%,纯度为98.75%。
44、esi-ms: m/z = 535.2(m+h) +。
45、1hnmr (400 mhz, dmso-d6) δ: 7.58~7.46 (m, 1h), 7.46~7.34 (m, 1h),7.39~ 7.24 (m, 2h), 6.29 (s, 1h), 4.75 (d, 1h), 3.90 (d, 2h), 3.71~3.60 (m,2h), 3.35 (m, 2h), 3.15~3.08 (m, 1h), 3.02 (t, 1h), 2.91 (t, 2h), 2.68 (m,2h), 2.39 (q, 1h), 2.31 (m, 2h), 1.74~1.37 (m, 10h)。
46、对比例1:2-氧代氯吡格雷(氯吡格雷活化过程中的代谢中间体)
47、
48、成都施贝康生物医药科技有限公司制备,ee=98.8%。
49、对比例2:维卡格雷(临床研究中)
50、
51、按照cn201010624329.7制备,纯度98.22%。
52、试验例1:抗血小板聚集作用研究
53、试验目的:评价并比较同摩尔给药后各化合物抗血小板聚集的治疗效果。
54、试验方法
55、(1)分组
56、将40只大鼠随机均匀分为4组:溶剂对照组、对比例1(2-氧代氯吡格雷)组、对比例2(维卡格雷)组、化合物1组。
57、(2)麻醉诱导
58、使用1.5%戊巴比妥钠对大鼠进行腹腔注射麻醉。
59、(3)固定
60、麻醉后的动物转移至操作台上,观察大鼠眼睑反射和痛觉反应,在眼睑反射反应及四肢和尾部痛觉反应消失后方可开始手术。
61、(4)大鼠腹主动脉取血
62、注射麻醉剂后,直到身体全身变软,方可把大鼠仰卧固定在手术台上,常规消毒后用手术剪刀沿腹正中线剪开腹腔,用小镊子轻轻扒开血管周围脂肪,再用棉球把覆盖在血管的多余脂肪擦净,直达清晰看清血管为止(腹主动脉位于脊柱上方;腹腔静脉血管比腹主动脉粗,颜色较深)。先固定血管,尽可避免血管移位,左手拇指和食指固定住血管两旁的脂肪及其它脏器,无名指按住血管进针点的上端,降低血压,可以避免喷血,右手持穿刺针,针尖斜面朝下,入针角度约 30度左右,朝向心端方向刺入,深度以5mm左右为宜,当针端回血后,将采血针另端刺入真空管中。进针后可以用止血夹夹住针头,可以避免麻醉不够挣扎导致血管被针头戳破。
63、(5)血小板制备
64、给药后3小时,戊巴比妥钠麻醉大鼠,然后腹主动脉采血,3.8%枸橼酸钠 1:9 抗凝,混匀后 200 g离心10 min,取上清为富血小板血浆(prp);余下血浆再以1600 g 离心15min,取上清为贫血小板血浆(ppp)。
65、(6)adp诱导血小板聚集
66、使用血小板聚集仪(美国海伦娜公司,型号:agg ram)测血小板聚集率:先取各通道待测prp所对应的ppp进行透光率校正,校正后取出ppp,再向各通道中放入已加入225 μl待测prp的比色皿,加入搅拌子,再加入adp(终浓度为 20μm),立即启动血小板聚集率检测。
67、试验结果
68、同摩尔给药后,各组血小板聚集率数据详见表1。
69、
70、结果显示:(1)溶剂对照组血小板聚集率(%)为:76.06±3.29;对比例1组、对比例2组、化合物1组的血小板聚集率(%)分别为31.32±6.07、39.32±12.38、20.06±5.32,各给药组与溶剂对照组比较,均具有显著的抑制adp诱导的大鼠血小板聚集作用(p均<0.01)。
71、(2)与对比例2组相比,化合物1组抑制adp诱导的大鼠血小板聚集作用有显著优势(p<0.01),具有统计学意义。
72、(3)从试验结果可以看出,同等剂量下单次静脉注射给药后,本发明中化合物1抑制adp诱导的大鼠血小板聚集作用明显优于对比例2化合物,也明显优于对比例1化合物。
73、试验例2:化合物对稳定过表达的herg通道电流的影响
74、试验样品
75、化合物1、对照化合物cisapride。
76、试验方法
77、采用手动膜片钳技术(herg安全性评价的金标准)来研究受试化合物1对herg钾通道的抑制作用,并评价其引发心室复极毒性的风险。
78、试验结果
79、本研究利用手动膜片钳技术检测受试化合物1对herg通道的阻断作用浓度效应关系,从而评价受试物对心脏herg钾通道抑制作用的风险。试验结果见表2。
80、
81、上述试验结果显示,本发明的化合物引发心室复极毒性的风险小,安全性高。
82、试验例3:药代动力学研究
83、试验目的
84、考察同摩尔剂量下,大鼠单次灌胃给药后,各个化合物的药代动力学特征,并对比主要药代动力学参数。
85、材料和方法
86、(1)受试物
87、对比例1(2-氧代氯吡格雷)、化合物1。
88、(2)给药制剂的配制
89、分别准确称取受试化合物于清洁给药容器中,加入适量solutol溶解,蜗旋震荡,加入纯水,超声,蜗旋震荡,直至化合物完全溶解;给药制剂均在给药当天新鲜配制。
90、(3)试验分组及给药情况
91、健康成年sd大鼠12只,灌胃给药。具体方案见表3。
92、
93、(4)试验方法
94、分组及禁食:sd大鼠,随机分组,每组6只,按表3灌胃给予相应化合物。
95、样品采集和处理:于给药前(0h)及给药后5 min、15 min、30 min、1 h、2 h、4 h、8h、24h于不同时间点采血0.2 ml,经edta-k2抗凝后4℃离心5 min,分离血浆于-80℃保存待测。
96、检测:采用lc/ms/ms法检测所有pk血浆样品中2-氧代氯吡格雷的血药浓度,并利用winnonlin7.2软件计算各个化合物的药代动力学参数。
97、试验结果
98、
99、等摩尔给药后,与对比例1化合物相比,化合物1的t1/2、cmax和auc0-last均有明显的延长和提高,具有统计学意义,p<0.01。说明化合物1在大鼠体内的吸收显著优于对比例1化合物,具有更优的药代动力学特征。
100、综上所述,本发明的化合物吸收更好,生物利用度高,预期更能充分发挥药效。
101、试验例4:溶解性研究
102、准确称量实施例化合物2和对比例2(维卡格雷)样品,于25~30℃条件下,用生理盐水及ph1.2的缓冲液各取1ml,再分别加入各化合物10 mg,测得溶解度数据见下表:本发明的化合物2具有优良的溶解度,可满足临床配制成有效浓度的注射液需求,而对比例2的溶解度极低,不符合注射液要求,若加入其它助溶剂,可能会带来潜在的安全性风险。
103、。
104、试验例5:影响因素试验研究
105、试验方法:分别称取化合物2和对比例2(维卡格雷)样品放置于称量瓶中,分别放置于高温(60℃)、高湿(rh80%)、光照(5000lux)条件。
106、化合物2的试验结果如下表所示:化合物2在高温、高湿、光照条件下10天,含量变化均较小,外观形状无明显变化,稳定性尚佳。
107、
108、而对比例2高温(60℃)放置两天,性状发生重大改变,从白色固体变为黄棕色油状物,说明对比例2高温条件下稳定性差。综上所述,本发明的化合物具有温、湿、光稳定性,且优于对比例2。
109、本领域的普通技术人员在不偏离本发明的精神的情况下,可对本发明化合物、组合物以及方法进行的多种修饰和变化,这些都属于与本发明相同或等同的范围。
- 还没有人留言评论。精彩留言会获得点赞!