一种抗真菌药物新的制备方法与流程
发明领域本发明属于化学药物合成领域,具体涉及一种抗真菌药物新的制备方法。
背景技术:
1、奥特康唑(oteseconazole)是mycoviapharmaceuticals公司开发的口服抗真菌,是fda批准的第一个治疗复发性外阴阴道念珠菌病(rvvc)的药物,奥特康唑用于在不具有生殖潜力的女性中,降低复发性外阴阴道假丝酵母菌病(rvvc)的复发几率,有效结合并抑制白色念球菌的cyp51(kd,<39nm);奥特康唑是一种唑类金属酶抑制剂,针对真菌甾醇14α脱甲基酶(cyp51),这种酶催化麦角甾醇生物合成途径的早期步骤,麦角甾醇是真菌细胞膜形成和完整性所需的甾醇。cyp51的抑制导致14-甲基化甾醇的积累,其中一些对真菌有毒性。通过包含四唑金属结合基团,奥特康唑对人cyp酶具有较低的亲和力,无明显作用。
2、复发性外阴阴道念珠菌病(rvvc)也被称为慢性酵母感染,是一种不同于外阴阴道念珠菌病(vvc)的疾病,定义为每年三次或三次以上有症状的酵母感染急性发作,主要症状包括阴道瘙痒、灼热、刺激和炎症。一些女性可能会经历异常阴道分泌物、性交疼痛或排尿疼痛,引起各种但往往是严重的不适和疼痛。近75%的成年女性一生中至少会感染一次酵母菌感染,其中大约一半会复发。在这些女性中,高达9%的人会患上rvvc。rvvc是一种慢性传染病,每年影响全球近1.38亿女性,仅美国就有600万女性,中国约有2900万rvvc患者。
3、奥特康唑的批准基于三项3期试验的积极结果,包括11个国家232个地点的875名患者。其中一项临床研究中93.3%在接受奥替康唑治疗的rvvc女性中,96.1%的女性在48周的维持期内没有复发,而接受安慰剂的女性分别为57.2%和60.6%(p<0.001)。另一项临床研究中,89.7%的女性接受奥替康唑治疗的rvvc清除了他们最初的酵母菌感染,并且在50周的维持期内没有复发,而接受氟康唑和安慰剂治疗的rvvc为57.1%(p<0.001)。
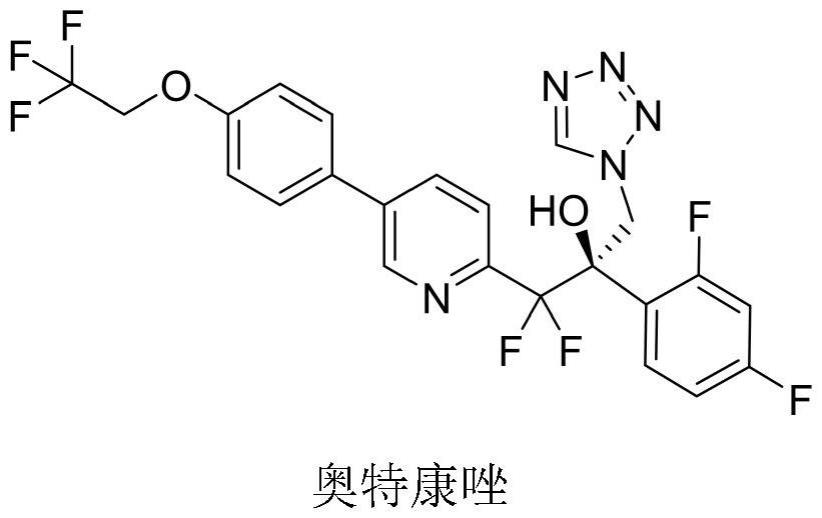
4、奥特康唑的中文化学名称:(r)-2-(2,4-二氟苯基)-1,1-二氟-3-(1h-四唑-1-基)-1-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-2-丙醇,分子式:c23h16f7n5o2,分子量:527.39,cas登记号:1340593-59-0,其化学结构式如下:
5、
6、现有技术文件专利cn201680061980报道了奥特康唑的合成路线1如下:
7、
8、现有技术文件专cn201180030668报道了奥特康唑的合成路线2如下:
9、
10、上述奥特康唑合成路线1不仅路线较长,且实施难度较大,其中步骤2涉及超低温反应,步骤3涉及剧毒品硝基甲烷使用,步骤4涉及加压氢化反应,步骤5涉及易爆危险品叠氮基三甲基硅烷使用,步骤6涉及贵重催化剂pd(dppf)cl2的使用,且整体收率较低,产业化意义不大。
11、上述奥特康唑合成路线2中步骤1使用致癌且易爆危险品重氮甲烷,步骤5通过拆分得到奥特康唑,不仅路线长,收率低,产业化意义同样不大。
12、为缓解奥特康唑的可及性问题,本发明提供一种奥特康唑的新方法,通过不对称合成高效率得到单一构型奥特康唑,该新方法在现有技术中尚无报道,虽然也涉及低温反应,但具有环境友好、路线短、收率高、成本低等优点。
技术实现思路
1、本发明提供一种抗真菌药物的制备方法,所述方法为:
2、
3、步骤1、(2,2,2-三氟乙氧基)苯硼酸频哪醇酯(式a化合物)与式b化合物经suzuki偶联反应得到2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物);
4、步骤2、2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物)与式d化合物偶联反应得到1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物);
5、步骤3、1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物)与式f化合物经不对称合成反应得到奥特康唑;
6、其中
7、式b化合物中x1选自i,br和cl,优选为br;
8、式d化合物中x2选自i,br和cl,优选为br;
9、式f化合物中x3选自i,br和cl,优选为br。
10、本发明最大的特点是设计了一条路线不同于现有技术文件专利cn201680061980和cn201180030668报道的奥特康唑新的合成路线,本发明仅通过3步反应制备得到奥特康唑,无剧毒和易爆物料使用,路线短,收率高,成本和环保优势明显。
11、本发明中,式b化合物中x1选自i,br和cl,其中氯代结构活性较弱,反应收率较低;碘代结构活性最强,但成本较高;而溴代结构2-(5-溴吡啶-2-基)-2,2-二氟乙酸乙酯不仅反应活性活性高,价廉易购,反应副产物少,收率较高,作为优选。
12、本发明中,式d化合物中x2选自i,br和cl,其中氯代结构活性较弱,反应收率较低;碘代结构活性最强,但成本较高;而溴代结构2,4-二氟溴苯不仅反应活性活性高,价廉易购,反应副产物少,收率较高,作为优选。
13、本发明中,式f化合物中x3选自i,br和cl,其中氯代结构活性较弱,反应收率较低;碘代结构活性最强,但成本较高;而溴代结构1-(2-溴乙基)-1h-四唑不仅反应活性活性高,价廉易购,反应副产物少,收率较高,作为优选。
14、本发明中步骤1suzuki偶联反应的催化剂为钯催化剂和无机碱催化剂,其中钯催化剂选自为四(三苯基膦)钯(0)、(1,1'-双(二苯基膦基)二茂铁)二氯化钯、双(三苯基膦)二氯化钯、双(三环己基膦)钯(0)或双(三-o-甲苯磷)钯(0),优选为四(三苯基膦)钯(0);无机碱选自碳酸铯,碳酸钾,碳酸钠或碳酸锂,优选碱性较大的碳酸铯或碳酸钾,因为溶剂体系中水含量较少,无机碱催化剂需进行粉碎或直接采用粉末状物料投料,以增加非均相反应的转化效率。
15、本发明中步骤1suzuki偶联反应中式b化合物的含有乙酯结构,为避免酯水解,仅加入痕量的水以保证反应顺利进行,同时了避免酯水解副产物的生成。另外为避免式b化合物酯交换副产物的生成,溶剂选择需避免乙醇以外的其它醇类溶剂使用。
16、本发明中步骤2suzuki偶联反应中在有机锂低温催化下进行,有机锂催化剂选自正丁基锂、叔丁基锂、仲丁基锂、正戊基锂、新戊基锂,其中正丁基锂活性最强,且成本较低,作为优选。
17、在本发明步骤2偶联反应中式c化合物、式d化合物的摩尔投料比为1:1~1.3:1~1.3,优选1:1.1~1.2:1.1~1.2,其中主物料式d化合物适当过量,能够使式c化合物反应完全,降低副产物产生,且过量物料容易通过后处理除去。
18、在本发明步骤3通过不对称合成反应高效制备单一构型的奥特康唑,是本专利最重要的特点,其中不对称反应催化剂选自(r)-1,1'-联二萘酚或3,3'-二取代芳基联萘酚,其中(r)-1,1'-联二萘酚选择性更强,反应效果重要更佳,且价格便宜,作为首选。另外本反应需加入格氏试剂异丙基氯化镁氯化锂催化偶联反应,该催化剂同时为路易斯酸;异丙基氯化镁氯化锂首先活化式f化合物,再在不对称合成催化剂(r)-1,1'-联二萘酚催化下与1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物)中羰基进行偶联反应,生成绝对单一r构型的奥特康唑,是本发明最大的亮点。
19、在本发明步骤3不对称合成反应中任何主物料的过量均会引起副产物的产生,稍过量的异丙基氯化镁氯化锂催化剂有利于反应进行,而不对称反应催化剂(r)-1,1'-联二萘酚仅需主物料的1/10当量即可,过量不仅增加成本,且导致副产物增多,经优化最终确认物料中式e化合物、式f化合物、异丙基氯化镁氯化锂及不对称反应催化剂(r)-1,1'-联二萘酚的摩尔投料比为1:0.9~1.1:1.0~1.5:0.08~0.12,优选1:0.95~1.05:1.1~1.3:0.09~0.11,能够保证反应顺利进行,同时将副产物控制在最低限度,并得的绝对单一r构型的奥特康唑,且成本较低。
20、本发明提供一条路线短,高收率,环境友好等优点的奥特康唑新的合成方法,相对现有技术文件优势明显,并可从其中获益。
21、具体实施方法:
22、下面将结合本发明实施例中,对本发明的实施例中的技术方案进行详细的说明,但如下实施例仅是用以理解本发明,而不能限制本发明,本发明可以由权利要求限定和覆盖的多种不同方式实施。
23、以下将结合本发明实施例1和10进一步说明奥特康唑新的合成方法及该方法的优势。
24、实施例1:2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物)的合成法1
25、
26、氮气保护下,将化合物对(2,2,2-三氟乙氧基)苯硼酸频哪醇酯(式a,151g,0.5mol)、2-(5-溴吡啶-2-基)-2,2-二氟乙酸乙酯(式b-i,140g,0.5mol)、四(三苯基膦)钯(0)(28.9g,25mmol)、粉状无水碳酸钾(79.5g,0.75mol),甲苯1l,无水乙醇200ml及水5ml置于三口烧瓶中,回流反应12h,冷却,加入1.2l水和乙酸乙酯500ml,搅拌分液,有机相再用1l水洗涤,有机相干燥,减压浓缩至约1/5体积,残余物冷却,过滤,滤饼烘干得到式c化合物(159g,收率85%),ms m/z 376.1(m+1)。1h nmr(300mhz d6-dmso):1.23(t,3h),4.18(q,2h),4.50(m,2h),7.01(d,2h),7.45(d,1h),7.73(d,2h),7.95(d,1h),8.79(d,1h)。
27、实施例2:2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物)的合成法2
28、
29、氮气保护下,将化合物对(2,2,2-三氟乙氧基)苯硼酸频哪醇酯(式a,75.5g,0.25mol)、2-(5-溴吡啶-2-基)-2,2-二氟乙酸乙酯(式b-i,70g,0.25mol)、(1,1'-双(二苯基膦基)二茂铁)二氯化钯(18.2g,25mmol)、粉状无水碳酸铯(122g,0.375mol),二甲基甲酰胺600ml、水2ml置于三口烧瓶中,控温80-90℃反应8h,冷却,加入1l水和乙酸乙酯1.2l,搅拌分液,有机相再用500ml水洗涤,有机相干燥,减压浓缩掉大部分溶剂,残余物加入正己烷200ml,回流溶解,冷却析晶,过滤,滤饼烘干得到式c化合物(67.6g,收率72%),ms m/z376.1(m+1)。
30、实施例3:2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物)的合成法3
31、
32、氮气保护下,将化合物对(2,2,2-三氟乙氧基)苯硼酸频哪醇酯(式a,60.4g,0.2mol)、2-(5-碘吡啶-2-基)-2,2-二氟乙酸乙酯(式b-i,65.4g,0.2mol)、四(三苯基膦)钯(0)(11.6g,10mmol)、粉状无水碳酸钾(31.8g,0.3mol),甲苯400ml,无水乙醇80ml及水2ml置于三口烧瓶中,回流反应6h,冷却,加入480ml水和乙酸乙酯200ml,搅拌分液,有机相再用400ml水洗涤,有机相干燥,减压浓缩至约1/5体积,残余物冷却,过滤,滤饼烘干得到式c化合物(75.1g,收率87%),ms m/z 376.1(m+1)。
33、实施例4:2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c化合物)的合成法4
34、
35、氮气保护下,将化合物对(2,2,2-三氟乙氧基)苯硼酸频哪醇酯(式a,30.2g,0.1mol)、2-(5-氯吡啶-2-基)-2,2-二氟乙酸乙酯(式b-i,23.6g,0.1mol)、四(三苯基膦)钯(0)(11.6g,10mmol)、粉状无水碳酸铯(65.2g,0.2mol),甲苯200ml,无水乙醇40ml及水1ml置于三口烧瓶中,回流反应18h,冷却,加入240ml水和乙酸乙酯100ml,搅拌分液,有机相再用200ml水洗涤,有机相干燥,减压浓缩至干,残余物加入乙酸乙酯100ml和正己烷150ml,加热溶解,冷却析晶,过滤,滤饼烘干得到式c化合物(21.8g,收率58%),ms m/z 376.1(m+1)。
36、实施例5:1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物)的合成法1
37、
38、氩气氛围下,将四氢呋喃(100ml、厂家为巴斯夫)和2,4-二氟溴苯(式d-i,44.4g,0.23mol)至1000ml干燥三口瓶中,冷却至-75~-70℃,控温-75~-55℃滴加n-buli(2.5m的正己烷溶液,92ml,0.23mol),滴毕,搅拌冷却至-75~-70℃,控温-75~-65℃滴加2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c,75g,0.2mol)的150ml的四氢呋喃溶液,滴加完毕,继续控温搅拌1h,滴加1m的盐酸溶液(250ml)猝灭反应,再用饱和碳酸钠溶液调ph 7-8,加入甲基叔丁基醚萃取两次(每次300ml),合并有机相,用纯化水300ml洗涤,有机相用无水mgso4干燥,过滤,有机相回流3-4h后,减压浓缩至干,残余物加入正己烷析出固体,过滤,滤饼用无水乙醇-正己烷(100ml/300ml)重结晶得到式e化合物(66.4g,收率75%,纯度98.8%),ms m/z 444.1(m+1),1h nmr(300mhz d6-dmso):5.14(d,2h),5.70(s,1h),6.91–6.95(m,1h),7.21(d,1h),7.23–7.25(d,2h),7.23-7.29(m,1h),7.30(d,1h),7.53(d,1h),7.83(d,2h),8.20–8.22(m,1h),8.90(d,1h)。
39、实施例6:1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物)的合成法2
40、
41、氩气氛围下,将四氢呋喃(50ml、厂家为巴斯夫)和2,4-二氟碘苯(式d-ii,26.4g,0.11mol)至500ml干燥三口瓶中,冷却至-75~-70℃,控温-75~-55℃滴加n-buli(2.5m的正己烷溶液,44ml,0.11mol),滴毕,搅拌冷却至-75~-70℃,控温-75~-65℃滴加2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c,37.5g,0.1mol)的80ml的四氢呋喃溶液,滴加完毕继续控温搅拌0.5h,滴加1m的盐酸溶液(125ml)猝灭反应,再用饱和碳酸钠溶液调ph 7-8,加入甲基叔丁基醚萃取两次(每次150ml),合并有机相,用纯化水150ml洗涤,有机相用无水mgso4干燥,过滤,有机相回流3h后,减压浓缩至干,残余物加入正己烷析出固体,过滤,滤饼用无水乙醇-正己烷(50ml/150ml)重结晶得到式e化合物(35.9g,收率81%,纯度99.1%),ms m/z 444.1(m+1)。
42、实施例7:1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e化合物)的合成法3
43、
44、氩气氛围下,将四氢呋喃(50ml、厂家为巴斯夫)和2,4-二氟氯苯(式d-ii,26.4g,0.13mol)至200ml干燥三口瓶中,冷却至-75~-70℃,控温-75~-55℃滴加n-buli(2.5m的正己烷溶液,23ml,57.5mmol),滴毕,搅拌冷却至-75~-70℃,控温-75~-65℃滴加2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)乙酸乙酯(式c,18.8g,50mmol)的40ml的四氢呋喃溶液,滴加完毕继续控温搅拌1h,滴加1m的盐酸溶液(70ml)猝灭反应,再用饱和碳酸钠溶液调ph 7-8,加入甲基叔丁基醚萃取两次(每次80ml),合并有机相,用纯化水80ml洗涤,有机相用无水mgso4干燥,过滤,有机相回流3h后,减压浓缩至干,残余物加入正己烷析出固体,过滤,滤饼用无水乙醇-正己烷(30ml/70ml)重结晶得到式e化合物(16.0g,收率72%,纯度97.5%),ms m/z 444.1(m+1)。
45、实施例8:奥特康唑的合成法1
46、
47、氩气氛围下,将1-(2-溴乙基)-1h-四唑(式f-i,17.7g,0.1mol)和四氢呋喃90ml加入至500ml干燥的三口瓶中,冷却至-35℃,控温-35~-15℃滴加1.3m异丙基氯化镁氯化锂的四氢呋喃溶液(110ml,0.12mol)。加毕,继续控温-25℃~-15℃搅拌反应1h。冷却至-25℃,控温-25~-15℃滴加1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e,44.3g,0.1mol)和(r)-1,1'-联二萘酚(2.9g,10mmol)的四氢呋喃(150ml)混合溶液,加毕,控温-25℃~-15℃反应1h,停止反应,向反应液中加入10%柠檬酸水溶液(200ml),用饱和碳酸钠溶液调ph至7-8,加入乙酸乙酯萃取三次(每次250ml),合并有机相,再用饱和食盐水300ml洗涤有机相,有机相无水硫酸钠干燥,过滤,滤液减压浓缩至干,残余物加入正己烷200ml搅拌分散,过滤,滤饼用无水乙醇200ml重结晶得到标题产物奥特康唑纯品(41.1g,收率78%,化学纯度98.6%,ee值98.2%),ms m/z 528.1(m+1),1hnmr(300mhz d6-dmso):4.86(q,2h),5.11(d,2h),5.68(s,1h),6.93–6.89(m,1h),7.19(d,1h),7.23–7.21(d,2h),7.27–7.21(m,1h),7.29(d,1h),7.52(d,1h),7.80(d,2h),8.22–8.20(m,1h),8.93(d,1h),9.15(s,1h)。
48、实施例9:奥特康唑的合成法2
49、
50、氩气氛围下,将1-(2-碘乙基)-1h-四唑(式f-ii,11.2g,50mmol)和四氢呋喃90ml加入至250ml干燥的三口瓶中,冷却至-35℃,控温-35~-15℃滴加1.3m异丙基氯化镁氯化锂的四氢呋喃溶液(55ml,60mmmol)。加毕,继续控温-25℃~-15℃搅拌反应0.5h。冷却至-25℃,控温-25~-15℃滴加1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e,22.2g,50mmol)和(r)-1,1'-联二萘酚(1.5g,5mmol)的四氢呋喃(80ml)混合溶液,加毕,控温-25℃~-15℃反应0.5h,停止反应,向反应液中加入10%柠檬酸水溶液(100ml),用饱和碳酸钠溶液调ph至7-8,加入乙酸乙酯萃取三次(每次120ml),合并有机相,再用饱和食盐水150ml洗涤有机相,有机相无水硫酸钠干燥,过滤,滤液减压浓缩至干,残余物加入正己烷100ml搅拌分散,过滤,滤饼用无水乙醇100ml重结晶得到标题产物奥特康唑纯品(21.1g,收率80%,化学纯度99.1%,ee值98.5%),ms m/z 528.1(m+1)。
51、实施例10:奥特康唑的合成法3
52、
53、氩气氛围下,将1-(2-氯乙基)-1h-四唑(式f-iii,29.1g,0.22mol)和四氢呋喃180ml加入至1l干燥的三口瓶中,冷却至-35℃,控温-35~-15℃滴加1.3m异丙基氯化镁氯化锂的四氢呋喃溶液(138ml,0.26mol)。加毕,继续控温-25℃~-15℃搅拌反应1.5h。冷却至-25℃,控温-25~-15℃滴加1-(2,4-二氟苯基)-2,2-二氟-2-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)-乙酮(式e,88.6g,0.2mol)和(r)-1,1'-联二萘酚(5.8g,20mmol)的四氢呋喃(300ml)混合溶液,加毕,控温-25℃~-15℃反应1.5h,停止反应,向反应液中加入10%柠檬酸水溶液(400ml),用饱和碳酸钠溶液调ph至7-8,用乙酸乙酯萃取三次(每次500ml),合并有机相,再用饱和食盐水600ml洗涤有机相,有机相无水硫酸钠干燥,过滤,滤液减压浓缩至干,残余物加入正己烷400ml搅拌分散,过滤,滤饼用无水乙醇300ml重结晶两次得到标题产物奥特康唑纯品(71.7g,收率68%,化学纯度97.6%,ee值97.2%),ms m/z528.1(m+1)。
54、本发明实施例1-10与对比例所提供的奥特康唑的合成方法相比较,路线更短,收率更高,减少了对生产操作人员的健康损害,环境更加友好。
55、以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!