一种艾滋病治疗药物来那卡帕韦中间体化合物的制备方法与流程
发明领域本发明属于化学药物领域,具体涉及一种艾滋病治疗药物来那卡帕韦的中间体化合物的制备方法。
背景技术:
1、获得性免疫缺陷综合征(acquired immunodeficiency syndrome,aids)(简称艾滋病)是由人类免疫缺陷病毒(human immunodefciency virus,hiv)引起的重大疾病。hiv可分为2种亚型:hiv-1和hiv-2,其中1型是目前全球流行的主要毒株,2型仅在西非流行。根据联合国艾滋病规划署《2020全球艾滋病防治进展报告》显示,目前世界上约有3800万艾滋病病毒感染者,2019年仍有69万人死于艾滋病相关疾病。但随着联合抗逆转录病毒疗法(combination antiretroviral therapy,cart)的发展,艾滋病已经逐渐从一种致命的大流行病转变为一种可控制的慢性疾病,艾滋病患者在使用cart治疗后可将自身hiv病毒载量保持在检测水平以下(<50copies/ml)并显著降低发病率及死亡率。虽然cart在对抗艾滋病上取得了卓越的成绩,但不可避免的多药耐药性及药物的不良反应仍严重限制cart的治疗效果。
2、来那卡帕韦(lenacapavir)是gilead sciences(吉利德科学公司)开发的一种人类免疫缺陷病毒1型(hiv-1)衣壳抑制剂,与其他抗逆转录病毒药物联合使用,适用于治疗经历过大量治疗且多重耐药hiv-1感染失败的成人的hiv-1感染。
3、来那卡帕韦是一种多级选择性hiv-1衣壳功能抑制剂,可直接结合六聚体中衣壳蛋白(p24)亚基之间的界面。表面等离子共振传感图显示来那卡韦与交联的野生型衣壳六聚体呈剂量依赖性和可饱和结合,平衡结合常数(kd)为1.4nm。来那卡帕韦通过干扰病毒生命周期的多个基本步骤来抑制hiv-1复制,包括衣壳介导的hiv-1前病毒dna核摄取(通过阻断与衣壳结合的核输入蛋白)、病毒组装和释放(通过干扰gag/gag-pol发挥作用,减少衣壳蛋白亚基的产生)和衣壳核心形成(通过破坏衣壳亚基结合的速率,导致畸形的衣壳)。
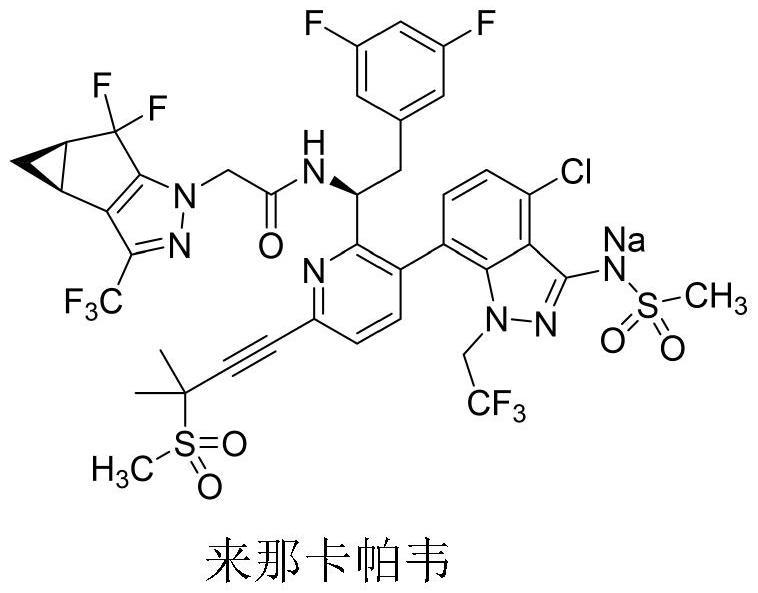
4、来那卡帕韦的中文化学名称:((4-氯-7-(2-((s)-1-(2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙[3,4]环戊[1,2-c]吡唑-1-基)乙酰氨基)-2-(3,5-二氟苯基)乙基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡啶-3-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)(甲基磺酰基)酰胺钠,分子式:c39h31clf10n7nao5s2,分子量:990.3,cas登记号:2283356-12-5,其化学结构式如下:
5、
6、现有技术文件专利cn201980026177报道了来那卡帕韦的合成路线1如下:
7、
8、现有技术文件专利cn201880053065报道了来那卡帕韦的合成路线2如下:
9、
10、上述路线1和路线2均存在收率低的问题,本发明的目的是提供一种仅需2步反应得到来那卡帕韦中间体化合物1的新方法,该化合物1再经甲磺酰基水解得到来那卡帕韦。
11、来那卡帕韦新的合成方法具有路线短,操作安全简单,成本低等优点。
12、因此,本发明有助于缓解中国艾滋病治疗药物来那卡帕韦的可及性问题。
技术实现思路
1、本发明的目的在于克服现有技术的不足,提供一种来那卡帕韦中间体n-((s)-l-(3-(4-氯-3-(n-(甲基磺酰基)甲磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡唑-2-基)-2-(3,5-二氟苯基)乙基)-2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑-1-基)乙酰胺(化合物1)新的制备方法,该中间体再经甲磺酰基水解得到来那卡帕韦。该方法具有路线短,操作安全简单,成本低等优点,所述方法为:
2、
3、步骤1、式a化合物和n-(4-氯-7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)-n-(甲基磺酰基)甲磺酰胺(式b化合物)经suzuki偶联反应得到(s)-n-[1-(3-(4-氯-3-(n-(甲磺酰基)甲基磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)]-2-氯乙酰胺(式c化合物);
4、步骤2、(s)-n-[1-(3-(4-氯-3-(n-(甲磺酰基)甲基磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)]-2-氯乙酰胺(式c化合物)与5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑(式d化合物)反应得到来那卡帕韦中间体化合物1。
5、本发明路线不同于现有技术文件专利cn201980026177和cn201880053065报道的来那卡帕韦的合成路线,本发明仅通过2步反应制备得到来那卡帕韦中间体化合物i,路线短,收率高,成本和环保优势明显。该化合物1再经一步水解反应得到来那卡帕韦。
6、本发明中式a化合物中含有氯乙酰基基团,式b化合物含有双甲磺酰基基团,上述基团均为不稳定活性基团,提前引入上述基团进行suzuki偶联反应有导致副产物增多的风险,这可能也是现有技术文件cn201980026177和cn201880053065中,一般采取在suzuki偶联反应后引入双甲磺酰基基团和酰胺基团的原因;然而,值得庆幸的是,本发明中式a化合物和式b化合物提前引入上述基团,其中式a化合物中卤素x为比氯乙酰基结构中氯代结构活性更强的br或i,虽然在suzuki偶联反应中产生少量副产物,但通过本发明中suzuki反应条件控制和后处理纯化可将式c化合物中副产物控制在较低水平,且收率较高。以上路线设计使本发明显著区别于现有技术文件路线,使来那卡帕韦的合成路线变得更短,更具成本优势。
7、本发明中步骤1的反应在无机碱催化下进行,为避免起始原料式a酰胺键水解,同时也为避免起始原料式b和产物式c中甲磺酰基水解,反应溶剂必须为非水溶剂,选自二氧六环、无水乙醇、甲苯、乙腈、二甲基甲酰胺、二甲基亚砜或者上述两种或多种溶剂的混合,考虑到主物料和无机碱的溶解性问题以及suzuki偶联反应的溶剂要求,优选二氧六环与无水乙醇的混合溶剂,即可保证起始物料、产物及无机碱碳酸锂的溶解,同时可保证反应顺利进行,且易于分离除去。
8、在本发明步骤1suzuki偶联反应中钯催化剂选自四(三苯基膦)钯(0)、[1,1’-双(二苯基膦基)二茂铁]二氯化钯(ii)、双(三苯基膦)二氯化钯、双(三环己基膦)钯(0)、双(三-o-甲苯磷)钯(0),其中四(三苯基膦)钯(0)和[1,1’-双(二苯基膦基)二茂铁]二氯化钯(ii)反应效果最佳,作为优选。尤其是四(三苯基膦)钯(0)做催化剂时,后处理不经重结晶操作即可得到高纯度的式c化合物,且收率较高,而采取其他钯催化剂,如双(三环己基膦)钯(0),需提高催化剂用量并增加反应时间,后处理需重结晶操作才能得到高纯度的式c化合物,收率稍降低。
9、在本发明步骤1suzuki偶联反应中无机碱催化剂,为避免物料和产物的降解,必须选择能在非水溶剂二氧六环与无水乙醇的混合溶剂中溶解的无机碱,而na2co3,k2co3,cs2co3在上述溶剂中溶解性较差,只能选择溶解性较好的li2co3或k3po4,试验结果显示:无机碱li2co3在有机溶剂中溶解性更好,能够保证步骤1suzuki偶联反应的顺利进行,且反应副产物较少,作为优选。
10、在本发明步骤1suzuki偶联反应中任何主物料的过量均可能导致副产物的产生,且后处理较难纯化除去,而过量的无机碱催化剂容易造成起始物料和产物的降解,而钯催化剂不仅价格昂贵,且过量容易产生三苯基氧膦副产物较难分离除去,经优化确认物料式a、式b、钯催化剂及无机碱催化剂的摩尔投料比为1:0.9~1.1:0.03~0.08:0.9~1.1,优选1:0.95~1.05:0.04~0.06:0.95~1.05,能够保证反应顺利进行,可将副产物控制在最低限度,同时有效控制了成本。
11、在本发明步骤1suzuki偶联反应控温70-80℃搅拌12-16h,既能保证反应完全,同是副产物较少,为最佳反应条件。
12、在本发明步骤2c-n偶联反应中催化剂为有机碱,有机碱选自n,n-二异丙基乙胺、n,n-二异丙基甲胺、三乙胺、n,n-二乙基甲胺、n,n-二乙基丙胺、n,n-二丙基-1-丙胺,优选为碱性较强的n,n-二异丙基乙胺,能够大大缩短反应时间,且后处理不经重结晶操作即可得到高纯度的化合物i,而采用碱性稍小的三乙胺,反应时间延长,且副产物增多,需通过重结晶纯化才能得到高纯度的化合物i。
13、在本发明步骤2c-n偶联反应中化合物式c、式d及有机碱催化剂的摩尔投料比为1:0.9~1.1:1.0~1.5,优选1:0.95~1.05:1.1~1.3,并控温0-10℃搅拌反应2~4h,反应副产物最少。
14、本发明提供一条路线短,高收率,具产业化潜力的来那卡帕韦中间体化合物1的新的合成方法,相对现有技术文件优势明显,并可从其中获益。
15、具体实施方法:
16、下面将结合本发明实施例中,对本发明的实施例中的技术方案进行详细的说明,但如下实施例仅是用以理解本发明,而不能限制本发明,本发明可以由权利要求限定和覆盖的多种不同方式实施。
17、以下将结合本发明实施例1-7以及对比例实施例9-11,进一步说明来那卡帕韦中间体化合物1新的合成方法及该方法的优势。
18、实施例1:(s)-n-[1-(3-(4-氯-3-(n-(甲磺酰基)甲基磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)]-2-氯乙酰胺(式c化合物)的合成法1
19、
20、氩气氛围下,将(s)-n-[1-(3-碘-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基]-2-氯乙酰胺(式a-i,58.1g,0.1mol)、n-(4-氯-7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)-n-(甲基磺酰基)甲磺酰(式b,53.2g,0.1mol),四(三苯基膦)钯(0)(5.8g,5mmol),碳酸锂(7.4g,0.1mol)、二氧六环800ml及无水乙醇200ml置于干燥的三口烧瓶中,控温70-80℃反应12h,浓缩掉2/3溶剂,残余物冷却至室温,加入水600ml和二氯甲烷600ml,搅拌分液,有机相再用水500ml洗涤,无水硫酸钠30g干燥2h,过滤,滤液减压浓缩掉4/5溶剂,残余物加入正己烷500ml搅拌分散,过滤,滤饼40℃减压烘干,得到高纯度标题产物式c(74.7g,收率87%)。msm/z 858.1(m+1)。1hnmr(300mhz d6-dmso):δ1.66(s,6h),2.85(s,3h),2.96(s,6h),2.99,3.25(m,2h),3.71(s,2h),4.23(q,2h),5.27(t,1h),6.56-6.65(m,3h),7.06(d,1h),7.67-7.68(d,2h),8.12(d,1h)。
21、实施例2:式c化合物的合成法2
22、
23、氩气氛围下,将(s)-n-[1-(3-碘-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基]-2-氯乙酰胺(式a-i,43.6g,75mmol)、n-(4-氯-7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)-n-(甲基磺酰基)甲磺酰(式b,39.9g,75mmmol),[1,1’-双(二苯基膦基)二茂铁]二氯化钯(ii)(3.3g,4.5mmmol),k3po4(15.9g,75mmol)、二氧六环600ml及无水乙醇150ml置于干燥的三口烧瓶中,控温70-80℃反应15h,浓缩掉2/3溶剂,残余物冷却至室温,加入水400ml和二氯甲烷400ml,搅拌分液,有机相再用水400ml洗涤,无水硫酸钠25g干燥2h,过滤,滤液减压浓缩掉4/5溶剂,残余物加入无水乙醇400ml和正己烷400ml回流溶解,结晶,过滤,滤饼40℃减压烘干,得到高纯度标题产物式c(63.5g,收率74%)。
24、实施例3:式c化合物的合成法3
25、
26、氩气氛围下,将(s)-n-[1-(3-碘-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基]-2-氯乙酰胺(式a,29.0g,50mmol)、n-(4-氯-7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)-n-(甲基磺酰基)甲磺酰(式b,26.6g,50mmmol),双(三环己基膦)钯(0)(2.0g,3mmol),碳酸锂(3.7g,50mmol)、二氧六环400ml及无水乙醇100ml置于干燥的三口烧瓶中,控温70-80℃反应15h,浓缩掉2/3溶剂,残余物冷却至室温,加入水300ml和二氯甲烷300ml,搅拌分液,有机相再用水300ml洗涤,无水硫酸钠20g干燥2h,过滤,滤液减压浓缩掉4/5溶剂,残余物加入无水乙醇200ml和正己烷200ml,回流溶解,结晶,过滤,滤饼40℃减压烘干,得到高纯度标题产物式c(27.9g,收率65%)。
27、实施例4:式c化合物的合成法4
28、
29、氩气氛围下,将(s)-n-[1-(3-溴-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基]-2-氯乙酰胺(式a,53.4g,0.1mol)、n-(4-氯-7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1-(2,2,2-三氟乙基)-1h-吲唑-3-基)-n-(甲基磺酰基)甲磺酰(式b,53.2g,0.1mol),四(三苯基膦)钯(0)(5.8g,5mmol),碳酸锂(7.4g,0.1mol)、二氧六环800ml及无水乙醇200ml置于干燥的三口烧瓶中,控温70-80℃反应16h,浓缩掉2/3溶剂,残余物冷却至室温,加入水600ml和二氯甲烷600ml,搅拌分液,有机相再用水500ml洗涤,无水硫酸钠30g干燥2h,过滤,滤液减压浓缩掉4/5溶剂,残余物加入正己烷500ml搅拌分散,过滤,滤饼用乙醇-正己烷(1:1)混合溶剂重结晶2次,滤饼减压烘干,得到高纯度标题产物式c(51.5g,收率60%),ms m/z 858.1(m+1)。
30、实施例5:n-((s)-l-(3-(4-氯-3-(n-(甲基磺酰基)甲磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡唑-2-基)-2-(3,5-二氟苯基)乙基)-2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑-1-基)乙酰胺(化合物1)的合成法1
31、
32、将(s)-n-[1-(3-(4-氯-3-(n-(甲磺酰基)甲基磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)]-2-氯乙酰胺(式c,64.4g,75mmol)、5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑(式d,16.8g,75mmol)及无水乙腈300ml置于干燥的三口烧瓶中,冷却至0℃,加入n,n-二异丙基乙胺(11.6g,90mmmol),控温0-10℃反应2h,终止反应,加入500ml二氯甲烷和300ml水,搅拌分液,有机相再用400ml水洗,无水硫酸钠25g干燥2h,过滤,滤液减压浓缩掉4/5体积溶剂,残余物加入正己烷300ml,搅拌分散,过滤,滤饼减压烘干得到高纯度化合物1(70.6g,收率90%,纯度98.2%),ms m/z 1046.1(m+1)。1hnmr(300mhz,d6-dmso):δ0.81-10.3(m,2h),1.65(s,6h),2.18(m,1h),2.86(s,3h),2.95(s,6h),2.99,3.18,3.25(m,3h),4.24(q,2h),4.60(s,2h),5.25(t,1h),6.55-6.65(m,3h),7.04(d,1h),7.68-7.69(d,2h),8.13(d,1h)。
33、实施例6:化合物1的合成法2
34、将(s)-n-[1-(3-(4-氯-3-(n-(甲磺酰基)甲基磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)]-2-氯乙酰胺(式c,42.9g,50mmol)、5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑(式d,11.2g,50mmol),无水乙腈200ml置于干燥的三口烧瓶中,冷却至0℃,加入三乙胺(6.5g,65mmmol),控温0-10℃反应4h,终止反应,加入350ml二氯甲烷和200ml水,搅拌分液,有机相再用300ml水洗,无水硫酸钠20g干燥2h,过滤,滤液减压浓缩掉4/5体积溶剂,残余物加入甲醇300ml和正己烷300ml,回流溶解,结晶过滤,滤饼减压烘干得到高纯度化合物1(38.2g,收率73%,纯度98.8%)。
35、实施例7:来那卡帕韦的合成
36、
37、参考对比文件cn201980026177中实施例11中操作;
38、将化合物1(52.3g,50mmol)、1mol/l的naoh溶液150ml及2-甲基四氢呋喃500ml置于三口烧瓶中,控温30-40℃反应2h。分离水相,有机相中加入水150ml洗涤,分液;有机相减压浓缩至干,残余物加入500ml无水乙醇,继续减压浓缩至约200ml,控温30-40℃,加入300ml正庚烷,冷却至室温析晶12h,过滤,滤饼用无水乙醇-正庚烷(1:1)适量淋洗,减压烘干得到标题产物来那卡帕韦(40.6g,收率82%,纯度99.5%)。
39、对比例:
40、中国专利cn201980026177中【实施例9、10、11】中所涉及的来那卡帕韦的合成工艺如下:
41、对比例实施例9:n-((s)-1-(3-(3-氨基-4-氯-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)-2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑-1-基)乙酰胺(iii)的制备
42、
43、向反应器中加入iv(1.0g)、碳酸氢钾(0.43g,1.3当量)、二氯双(三环己基膦)钯(ii)(28mg,2.5mol%)、v-02(0.67g)、乙酸丁酯(7.3g)和水(2.1g)。将反应器惰性化,将混合物在约85℃(75-90℃)下搅拌直至反应完全。将混合物冷却至约40℃并通过硅藻土(0.52g)。用乙酸丁酯(1.8g)冲洗硅藻土饼。合并滤液和冲洗液,用溶于水(5.2g)中的n-乙酰基-l-半胱氨酸(0.31g)和在水中的氢氧化钠(5wt%,5.4g)的混合物洗涤该溶液两次。将有机物用水中的氯化钠(5wt%,11g)洗涤两次。将溶液共沸蒸馏到1-丙醇(3.3g)中。在约50℃下向丙醇溶液中加入甲磺酸(0.31g,2.25当量),并使用二丁醚(5.1g)使产物结晶。将浆液冷却至约10℃,过滤,用正丙醇在二丁醚中的5:1混合物(1.6g)洗涤滤饼,将固体干燥以提供iii-03。
44、在约20℃下将氢氧化钠水溶液(9.2g,0.2m,2.2当量)加入含有2-甲基四氢呋喃(8.3g)中的iii-03(1.0g)的反应器中。将两相混合物搅拌约15分钟,并除去水层。将有机层用2.0wt%的氯化钠水溶液(9.8g)洗涤四次并蒸馏以提供iii。
45、对比例实施例10:n-((s)-1-(3-(4-氯-3-(n-(甲基磺酰基)甲磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)-2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑-1-基)乙酰胺(ii)的制备
46、
47、在约10℃下将甲磺酰氯(0.32g,2.5当量)加至含有iii(1.0g)、三乙胺(0.69g,6.0当量)和methf(11g)的反应器中。将混合物在约10℃下搅拌直至反应完成。用水(6.4g)洗涤反应混合物约15分钟,并升温至约20℃。分离各层,用10wt%氯化钠水溶液(6.9g)洗涤有机层约15分钟。分离分层,将有机层直接用于下一步。
48、对比例实施例11:n-((s)-1-(3-(4-氯-3-(甲磺酰胺基)-1-(2,2,2-三氟乙基)-1h-吲唑-7-基)-6-(3-甲基-3-(甲基磺酰基)丁-1-炔-1-基)吡啶-2-基)-2-(3,5-二氟苯基)乙基)-2-((3bs,4ar)-5,5-二氟-3-(三氟甲基)-3b,4,4a,5-四氢-1h-环丙烯并[3,4]环戊二烯并[1,2-c]吡唑-1-基)乙酰胺(i,来那卡帕韦)的制备
49、
50、在约35℃下将氢氧化钠(1m,2.9g,3.0当量)加至含有ii(1.0g)和2-甲基四氢呋喃(8.4g)的反应器中。搅拌混合物直至反应被认为完全。将反应混合物调节至约20-40℃,并除去底层。用水(2.9g)洗涤有机层约15分钟,除去底层。将有机溶剂换成乙醇,并将溶液浓缩至约5体积,并将温度调节至约35℃。缓慢加入正庚烷(3.4g),并将混合物老化约12h。过滤收集固体,用乙醇/正庚烷(1:1)洗涤滤饼。将所得湿饼在真空下干燥以提供i-02。
51、在约20℃下将化合物i-02(1.0g)和冰乙酸(2.1g)合并并搅拌直至溶解。在约1小时内将所得溶液转移至含水(15g)的反应器中。将所得浆液进一步搅拌约1小时并过滤。用水(2×5g)洗涤湿饼,脱液,并在约60℃真空下干燥以提供i(来那卡帕韦)。
52、最后有必要说明的是,以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!