一种氟吡菌酰胺的制备方法与流程
本发明涉及一种氟吡菌酰胺的制备方法,属于有机化学。
背景技术:
1、氟吡菌酰胺是由拜耳发现并开发的苯甲酰胺类杀菌剂,为琥珀酸脱氢酶抑制剂(sdhi)。其化学名称为:n-{2-[3-氯-5-(三氟甲基)吡啶-2-基]乙基}-2-(三氟甲基)苯甲酰胺;分子式:c16h11clf6n2o;相对分子质量:396.76;cas登录号:658066-35-4。氟吡菌酰胺是优秀的杀菌剂和杀线虫剂,现已在全球60多个国家和地区登记和上市,用于70多种作物。该产品可高效防治许多病害,如灰霉病、白粉病、菌核病、大豆猝死综合症等,而且可用于防治许多线虫,如根结线虫、根腐线虫、穿孔线虫、毛刺线虫、刺线虫等,是sdhi类杀菌剂中首个提供杀线虫活性的化合物。氟吡菌酰胺毒性低,用量少,对环境友好。
2、德国拜耳作物科学公司2003年8月8日在中国申请化合物发明专利(专利号:038194716)。并于2012年11月5日在中国获得:96%氟吡菌酰胺原药农药登记(登记证号pd20121673)。
3、目前,氟吡菌酰胺在实际生产中,存在着生产路线长,成本控制较难,收率较低,产品品质较差,纯度和含量都较低等问题,为提高市场竞争力,需要找到一种生产路线短,成本低,收率高,产品纯度和含量高的制备工艺路线。氟吡菌酰胺的工艺路线主要有以下几种:
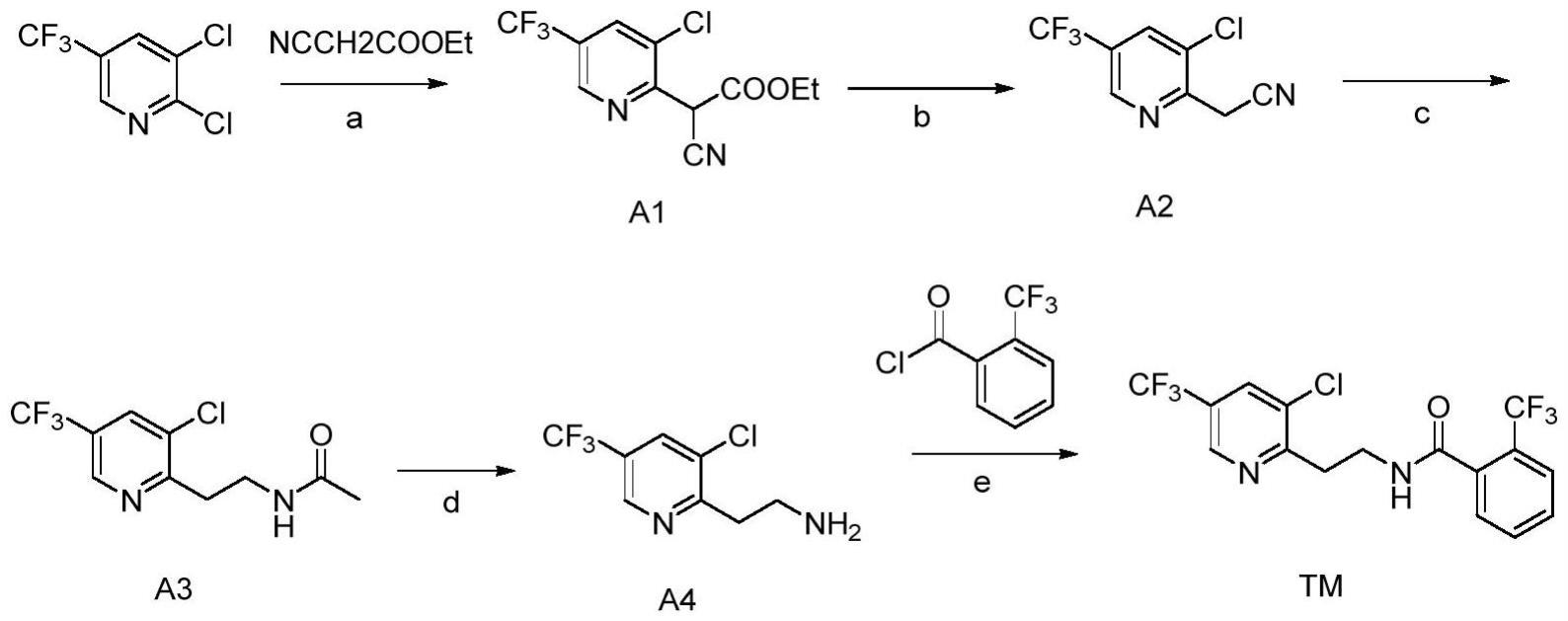
4、路线一:专利ep1674455公开了以2,3-二氯-5-三氟甲基吡啶和氰乙酸乙酯为原料,经过亲核取代、水解脱羧、催化氢化、水解制得3-氯-5-(三氟甲基)-2-(2-氨基乙基)吡啶a4,最后与中间体邻三氟甲基苯甲酰氯反应得到氟吡菌酰胺。该路线总收率低约44%,此外该路线中需要使用贵金属钯催化氢化还原,极大地提高了成本,同时该氢化步骤,较容易产生脱氯的副反应,生成难除去的杂质,不利于产业化生产。
5、
6、路线二:专利wo2018/114484公开了以邻三氟甲基苯甲酸为原料,经酰化、氨化、羟甲基化、酯化制成中间体b5;另以2,3-二氯-5-三氟甲基吡啶与丙二酸二乙酯为原料缩合制成中间体b1,然后将中间体b5与中间体b1进行缩合制成中间体b6,最后水解脱羧得到氟吡菌酰胺。该工艺路线,步骤多达7步,同时中间体b5与中间体b1进行缩合的反应条件较为剧烈,且总收率较低约49%,操作较复杂繁琐,提高了成本,不利于产业化生产。
7、
8、路线三:专利cn113620867a采用2,3-二氯三氟甲苯经过氟化、氰基取代、水解、羟甲基化、酯化制得中间体c6;另将2,3-二氯-5-三氟甲基吡啶与丙二酸二酯反应制得中间体c1;将中间体c6与中间体c1缩合后,再进行水解、脱羧,最后氢化脱氯制得氟吡菌酰胺,该路线长,副反应多,特别是该路线中需要使用贵金属钯催化氢化还原,极大地提高了成本,同时该氢化步骤,难以控制选择性脱氯的反应条件,容易生成难除去的杂质,不利于产业化生产,同时产物的总收率低,只有50.3%。
9、
10、基于上述原因,亟需一种制备氟吡菌酰胺的方法,以解决氟吡菌酰胺在存在着生产路线长,成本控制较难,收率较低,产品品质较差,纯度和含量都较低等问题,提高市场竞争力。
技术实现思路
1、本发明所要解决的技术问题为:提供一种具有高收率、高纯度、低成本的氟吡菌酰胺的制备方法。
2、本发明通过以下技术方案解决上述技术问题:
3、本发明提供了一种氟吡菌酰胺的制备方法,包括以下步骤:
4、步骤1、在溶剂1和碱的存在下,加入丙二酸二甲酯、2,3-二氯-5-三氟甲基吡啶进行反应,调节ph至4-6,萃取,得粗产物,再将粗产物加入到醇钾盐的醇溶剂中,进行成盐反应,得到2-[3-氯-5-(三氟甲基)吡啶基]丙二酸二甲酯钾盐,即中间体1;
5、步骤2、在溶剂2存在下,加入邻三氟甲基苯甲酰胺、水、碱、甲醛水溶液进行反应,反应结束后,得到中间体2;
6、步骤3、加入中间体2、溶剂3和氯化试剂进行反应,得到n-(氯甲基)-2-三氟甲基苯甲酰胺,即中间体3;
7、步骤4、加入溶剂4、中间体1、中间体3,反应结束后,得到中间体4;
8、步骤5、加入水、碱、溶剂5、中间体4,反应后除去溶剂、调节ph,继续反应,反应结束后,得到产物氟吡菌酰胺。
9、
10、优选地,所述的步骤1中,溶剂1选自n,n-二甲基乙酰胺和/或n,n-二甲基甲酰胺,调节ph所用的溶剂选自盐酸;所述的碱选自氢氧化钾;所述的醇溶剂选自叔丁醇、甲醇和异丙醇中的至少一种;醇钾盐选自叔丁醇钾和/或甲醇钾;2,3-二氯-5-三氟甲基吡啶与溶剂1的质量比为1:3.0-7.0;2,3-二氯-5-三氟甲基吡啶和碱的摩尔比为1:1.0-1.2;2,3-二氯-5-三氟甲基吡啶和丙二酸二甲酯的摩尔比为1:1.0-1.2;2,3-二氯-5-三氟甲基吡啶与醇钾盐的摩尔比为1:1.1-2.0;2,3-二氯-5-三氟甲基吡啶与醇溶剂的质量比为1:2.0-8.0;加入时的温度为25-40℃;反应温度为25-50℃,反应时间为2-5小时;成盐温度为10-40℃,成盐反应时间为0.5-3小时。
11、进一步优选地,所述的步骤1调ph后还包括后处理步骤:萃取,合并有机相,减压浓缩;所述的2,3-二氯-5-三氟甲基吡啶与萃取溶剂的质量比为1:3.0-8.0,萃取溶剂为甲基叔丁醚。
12、进一步优选地,所述的步骤1反应结束后还包括后处理步骤:过滤,过滤母液可套用,滤饼用醇溶剂淋洗后,烘干,所述的醇溶剂选自叔丁醇、甲醇和异丙醇中的至少一种。
13、优选地,所述的步骤2中,溶剂2选自1,2-二氯乙烷;碱选自碳酸钠;邻三氟甲基苯甲酰胺与水的质量比选自1:3.0-10.0;邻三氟甲基苯甲酰胺与碱的摩尔比选自1:0.02-0.1;邻三氟甲基苯甲酰胺与甲醛水溶液中甲醛的摩尔比为1:2.0-6.0;甲醛水溶液中甲醛的质量分数为25%-40%,邻三氟甲基苯甲酰胺与溶剂2的质量比为1:4.0-10.0;加入的温度为60-90℃;反应温度为70-100℃,反应时间为6-18小时。
14、进一步优选地,所述的步骤2反应结束后还包括后处理步骤:反应液萃取,有机相合并水洗,干燥,减压浓缩。
15、优选地,所述的步骤3中,溶剂3选自n,n-二甲基乙酰胺和/或二甲基亚砜;中间体2与氯化试剂的摩尔比为1:1.0-5.0;中间体2与溶剂3的质量比为1:2.0-6.0;反应的温度为5-60℃,反应时间为3-6小时;氯化试剂选自三甲基氯硅烷。
16、进一步优选地,所述的步骤3反应结束后还包括后处理步骤:高真空减压浓缩。
17、优选地,所述的步骤4中,溶剂4选自n,n-二甲基乙酰胺和/或二甲基亚砜;碱选自氢氧化钾;所述的中间体1与中间体3的摩尔比为1:1.0-1.2;中间体1与溶剂4的质量比为1:2.0-6.0;加入温度为40-60℃;反应的温度为45-65℃,反应时间为2-4小时。
18、进一步优选地,所述的步骤4反应结束后还包括后处理步骤:减压回收溶剂;向残余物中加入有机溶剂和水,萃取后分液,有机相用水洗涤,减压浓缩,重结晶;有机溶剂优选为n,n-二甲基乙酰胺和/或二甲基亚砜;萃取所用的溶剂优选为甲基叔丁基醚和/或1,2-二氯乙烷;重结晶所用的溶剂优选为甲醇、乙醇和水中的至少一种。
19、优选地,所述的步骤5中,所述的溶剂5选自甲醇;所述的碱选自氢氧化钾;中间体4与水的质量比为1:1.0-3.0;所述的中间体4与碱的摩尔比为1:3.5-6.0;所述的中间体4与溶剂5的质量比为1:1.0-3.0;反应的温度为25-45℃,反应时间为3-6小时;继续反应的温度为40-60℃,继续反应的时间为2-5小时。
20、进一步优选地,所述的步骤5反应结束后还包括后处理步骤:过滤,固体用水洗涤后,再次过滤,干燥、重结晶,过滤,干燥;重结晶所用的溶剂为甲醇和/或水;中间体4与重结晶所用的溶剂的质量比为1:2.0-6.0。
21、本发明与现有技术相比,具有如下有益效果:
22、1、本发明步骤1中,以2,3-二氯-5-三氟甲基吡啶为原料,通过控制温度等工艺条件,通过发生缩合反应并成盐的方法,高收率的得到了2-[3-氯-5-(三氟甲基)吡啶基]丙二酸二甲酯钾盐,同时成盐所用的醇钾盐的醇溶液可套用于下一批次成盐,原料利用率高,减少对环境的污染;分离效率高,收率高,纯度高,反应时间短,反应条件温和,适合大生产。
23、2、本发明步骤3中,通过大量实验,发现三甲基氯硅烷,作为氯化试剂,能高收率的得到n-(氯甲基)-2-三氟甲基苯甲酰胺即中间体3,该制备工艺和后处理工艺简单,产物无需纯化可用于下一步反应;原料综合利用率高,该方法能大大提高中间体3的纯度和品质,有利于提高后续步骤的中间体4产率和简化纯化工艺。
24、3、本发明步骤4中,反应时间短,反应条件温和,能高收率的得到中间体4,反应的选择性提高,抑制了杂质的生成,通过简单重结晶即可得到高纯度的中间体4;同时本发明步骤5中,通过控制水和碱的量以及反应温度,使水解和脱羧反应可以一锅法进行,大幅降低三废量,降低了生产时间和后处理工艺;通过简单重结晶即可高收率的得到高纯度的产物氟吡菌酰胺。该方法产物氟吡菌酰胺纯度≥99.0%,质量含量≥98.3%,五步总收率≥68.2%。
- 还没有人留言评论。精彩留言会获得点赞!