基因组多重结构变异的检测方法及其应用
本发明涉及分子生物学技术应用领域,具体涉及一种基因组多重结构变异的检测方法及其应用。
背景技术:
1、在肿瘤的基因组中存在有大量的基因组变异,包括基因组结构变异(structuralvariation,sv)、染色体拷贝数异常(copy number variation,cnv)、染色体内均匀扩增(homogenously staining regions,hsr)、染色体外dna环状扩增(extrachromosomal dna,ecdna),以及单核苷酸突变(single nucleotide variations)和小范围的碱基缺失(<5bp,indel)等。这些变异对肿瘤的增殖,转移,与耐药有着密切的联系。此外,在不孕不育患者的基因组中,存在有大量的平衡易位,这些易位携带者通常表型和智力正常,但在生殖细胞减数分裂时可产生各种不平衡重排的配子,造成不孕不育、流产、死产、死胎以及生育染色体异常的多发性畸形儿等。
2、全基因组高通量测序,能够高效的检测单碱基突变及小的indels。但是由于测序读长的限制,大范围的基因组结构变异,包括染色体内和染色体间的倒位,平衡易位,传统的高通量测序方法,难以效的检测。
3、为了对大尺度的结构变异有更准确的检测,发展了大量的基于短序列、单分子长片段、以及光学成像的三代测序技术,如:linked reads,strand-seq,pacificbiosciences(pacbio),and oxford nanopore technologies(ont),bionano等。这些技术的产生和发展对检测大范围的基因组结构变异有积极的推动作用,但是这些技术有一个共同特点:成本昂贵,检测周期长,依赖复杂的仪器设备,因此目前这些技术都没有在临床上应用起来。
4、hi-c已经被证明了,在检测染色体结构变异,倒位易位,有着非常强大的能力,但是标准的hi-c过程,步骤十分的繁琐,起始细胞量要求大,其中的生物素标记及磁珠回收的步骤,试剂成本昂贵,且周期长。由于hi-c只捕获了连接节点处的染色体互作信息,因此,离酶切位点较远处的单核苷酸变异,以及indel,难以被检测到。因此hi-c只适合大尺度结构变异的检测,而对小的结构变异检测的能力较差。此外,针对snp,indel,和cnv,主要采取的是全基因组测序(wgs)与全外显子测序(wes)的策略。而对于大尺度的结构变异,例如sv,hsr,ecdna等,主要采用的是主要采用的是三代测序+wgs+hi-c联合测序的策略,但是由于高昂的文库构建与测序成本,目前该方法没有在临床上进行推广与运用。目前临床上亟需一种多类型基因组结构变异的检测技术,以满足精准诊断与精准用药的需求。
5、wgs和wes是检测单碱基突变与小的碱基插入与缺失(<5bp)的金标准,但由于其没有基因组的空间互作信息,因此对大范围的基因组结构变异(sv)检测能力较差。hi-c技术自发明以来在染色体结构变异(sv)的检测中展现出了出色的潜能。通过绘制全基因组染色质互作图谱,hi-c可以有效的鉴定染色体倒位、易位、缺失等各种类型的基因组结构变异。但是由于hi-c只能捕获连接结点处的染色体互作信息,因此覆盖度较低,对小范围的结构变异以及染色体拷贝数变异的检测能力较差。目前还没有一项技术能够全面的对多类型结构变异进行检测。此外,由于三代测序(包括pacbio,nanopore,和bionano)的成本昂贵、起始dna用量大,文库制备复杂,不适合临床应用。
技术实现思路
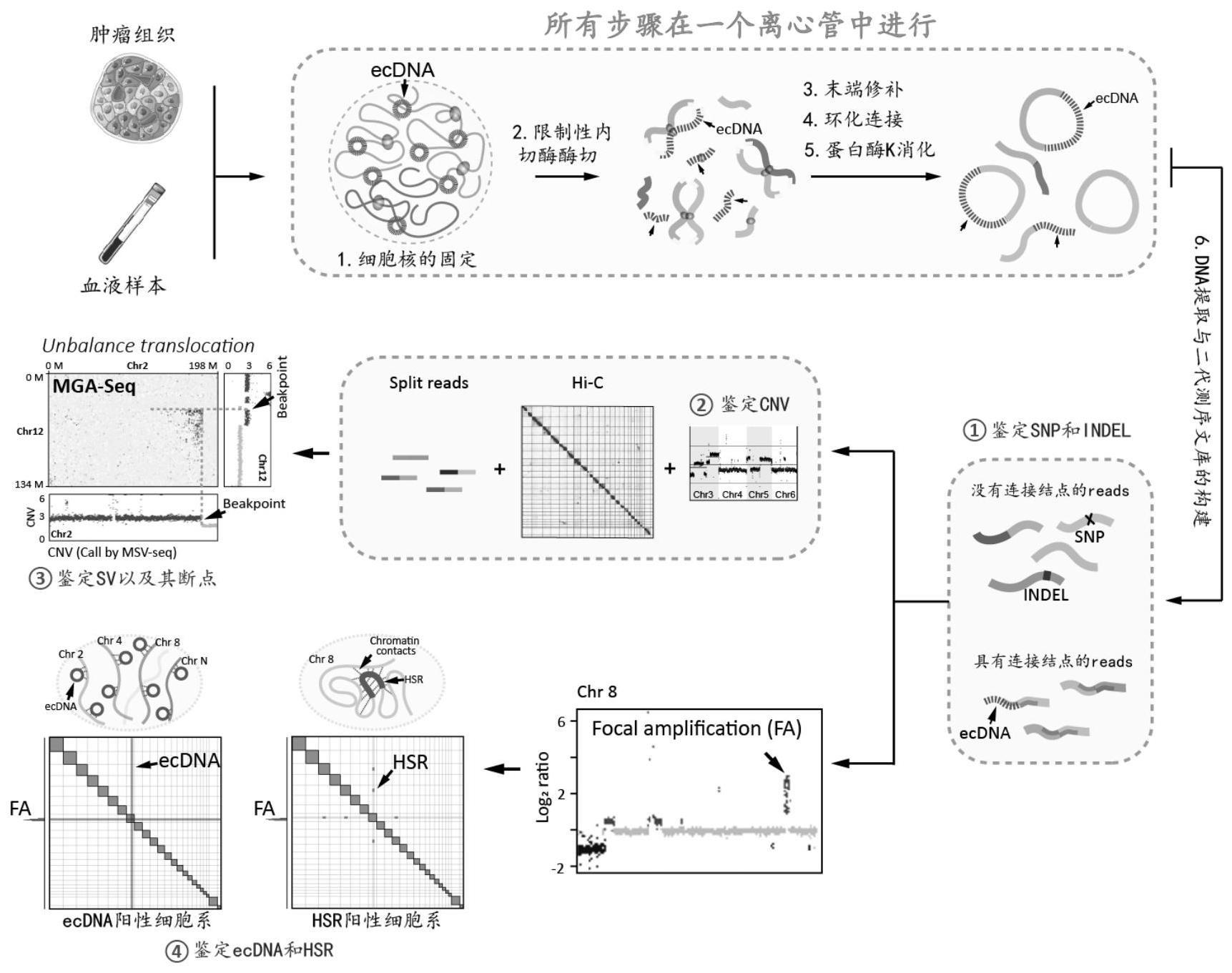
1、本发明针对传统的基于三代测序的基因组结构变异检测技术,如linked reads,strand-seq,pacific biosciences(pacbio),oxford nanopore technologies(ont),以及bionano等技术具有的共同的缺陷:成本昂贵,检测周期长,依赖复杂的仪器设备;提供了一种基因组多重结构变异的检测方法(multiple genetic abnormality sequencing(mga-seq));该方法利用了反应缓冲液的通用性,在一个离心管中即可完成mga-seq测序文库的构建。通过高通量测序与分析,可以实现对包括sv,cnv,hsr,ecdna,snp,和indel在内的多种类型基因组结构异常进行有效的检测。
2、为实现上述目的,本发明所设计的技术方案如下:
3、本发明提供了一种基因组多重结构变异的检测方法,包括以下步骤:
4、1)待检测样本用pbs缓冲液润洗,然后加入胰酶进行消化;将终止消化的样本细胞转移到离心管中,离心收集沉淀,pbs缓冲液洗涤沉淀,向沉淀加入pbs缓冲液制作细胞悬浮液;
5、2)向细胞悬浮液中加入甲醛固定,离心收集沉淀,pbs缓冲液清洗沉淀;向沉淀加入pbs缓冲液制作细胞核悬浮液;
6、3)向细胞核悬浮液中加入sds和sigme蛋白酶抑制剂进行室温裂解;离心,去上清,将细胞核用pbs缓冲液清洗一遍;
7、4)将细胞核悬浮于ddh2o中,然后配置含有nebuffer2.1的酶切反应体系,震荡酶切,得到酶切产物;
8、5)将酶切产物进行震荡反应末端补平,得到末端补平产物;
9、6)将末端补平产物配置酶连反应体系,慢旋转连接,得到酶连产物;
10、7)酶连产物进行消化,消化结束后,用zymo pcr产物纯化试剂盒进行dna提取;
11、8)用vazyme的dna建库试剂盒将其构建成高通量测序文库进行高通量测序;
12、9)数据拆分:利用seqkit(seqkit是一款专门处理fsata/q序列文件的软件)过滤含有hi-c ligation junction的reads对,得到拆分后的数据(拆分后的数据用于除ecdna、hsr之外其他类型变异的检测);
13、10)突变和缺失(snp和indel)检测:利用gatk软件包对拆分后数据进行snp和indel的鉴定;
14、11)结构变异(sv,structural variation)检测:使用两种结构变异的检测方法delly2与gridss对拆分后数据进行结构变异的检测与过滤,再用survivor进行结果整合;
15、12)拷贝数变异(cnv)检测:使用bic-seq2软件对拆分后数据进行cnv检测;
16、13)ecdna的检测:使用软件juicer对拆分前的完整数据进行染色体三维结构交互矩阵构建,结合cnv检测结果与三维结构信息,使用gwifa(基于全基因组范围的三维结构交互波动,判别ecdna或者hsr)方法对ecdna和hsr进行鉴定,完成基因组多重结构变异的检测。
17、进一步地,所述步骤1)中,待检测样本为肿瘤组织或抗凝全血。
18、再进一步地,所述步骤1)中,细胞悬浮液具体制作方法如下:
19、a.将待检测样本用1ml pbs缓冲液润洗,然后向t25的细胞瓶加入0.5ml的胰酶,温度为37℃消化3min,消化完后,用1ml含血清的培养基终止;
20、b.将细胞转移到15ml离心管中,离心收集沉淀,将沉淀用1ml的pbs缓冲液洗涤一遍;最后用5ml pbs缓冲液制作细胞悬浮液。
21、再进一步地,所述步骤2)中,细胞悬浮液中,甲醛的终浓度为0.5%。
22、再进一步地,所述步骤3)中,裂解时间为5min;裂解过程中,每1ml的细胞核悬浮液中,使用20ul的质量分数为10%的sds和4ul的sigme蛋白酶抑制剂。
23、再进一步地,所述步骤4)中,配置酶切反应体系如下:
24、
25、
26、震荡条件为:温度为37℃,转速为800rad/min,震荡酶切2h。
27、酶切反应体系还可能根据实际情况选择:clai、psti-hf、mboi、dpnii、msei。
28、再进一步地,所述步骤5)中,末端补平的反应体系为:
29、 酶切产物 200ul dntp mix(10mm each) 5ul dna polymerase i,large(klenow)fragment(neb,m0210) 5ul 合计 210ul
30、37℃,转速为800转/min震荡反应1h。
31、再进一步地,所述步骤6)中,酶连反应体系为:
32、 末端补平产物 210ul t4 dna ligase buffer 30ul t4 dna ligase 20ul <![cdata[h<sub>2</sub>o]]> 40ul 合计 300ul。
33、再进一步地,所述步骤7)中,消化的反应体系如下:
34、 酶连产物 300ul 质量分数为10%的sds 25ul 蛋白酶k 25ul 合计 350ul
35、本发明还提供了一种上述方法在分子分型和筛选中的应用。
36、本发明的有益效果:
37、本发明的基因组多重结构变异的检测方法,通过利用反应缓冲液的通用性,只需要在一个离心管中进行固定、酶切、末端补平、邻近连接、以及dna提取即可完成文库的构建,简化了文库构建的步骤,提高了文库构建的成功率,可以在一种测序文库中实现对snvs,indels(<5bp),svs,以及ecdna和hsr等各种类型的基因组结构变异进行精准检测,大大降低了文库构建成本,避免了由于更换反应缓冲液反复离心导致的样本损失,对辅助生殖中优质囊胚的筛选,肿瘤精准诊断等领域有巨大的应用价值。
- 还没有人留言评论。精彩留言会获得点赞!