4-(3-吲哚基)-1,3-噻唑类化合物及其制备方法和应用
本发明涉及药物化学,具体涉及一种4-(3-吲哚基)-1,3-噻唑类化合物及其制备方法和在制备组蛋白赖氨酸特异性去甲基化酶1(lsd1)抑制剂中的应用。
背景技术:
1、lsd1是第一个被发现的组蛋白去甲基化酶,通过黄素腺嘌呤二核苷酸(fad)依赖型机制特异性脱去单甲基化和二甲基化h3k4和h3k9位点上的甲基基团,从而调节基因表达和转录活性。研究表明:lsd1在多种肿瘤细胞中过表达,可通过组蛋白的去甲基化作用激活或抑制的染色质结构域从而调控基因的表达,并且通过影响细胞增殖、分化中所必需因子的表达,从而调控肿瘤的发生与发展(biomolecules,2022,12,462)。此外,lsd1与其他疾病如病毒感染、中枢神经系统疾病、心脑血管疾病等发生发展亦密切相关(epigenomics,2016,8,1103–1116;journal of hematology&oncology,2019,12,129)。
2、目前已有较多的lsd1抑制剂报道,但尚无用于肿瘤治疗的上市lsd1抑制剂(journal of medicinal chemistry,2023,66,71–94)。lsd1抑制剂根据作用方式分为共价抑制剂和可逆抑制剂,处于临床研究阶段的lsd1抑制剂多数为苯环丙胺类共价抑制剂,这类lsd1共价抑制剂不可逆共价结合fad,对以fad为辅因子的多种靶标均表现出高亲和力,具有一定的潜在毒性。可逆抑制剂是通过非共价的方式与fad结合,在安全性方面具有一定的优势,其中cc-90011和sp-2577已进入i/ii期临床用于肿瘤治疗(journal of medicinalchemistry,2020,63,14522–14529;journal of clinical oncology,2021,39,11514-11514)。此外,lsd1抑制剂与其他药物联用治疗肿瘤也已经进入临床研究阶段。虽然可逆抑制剂在安全性方面有潜在优势,但其结构类型相对有限。
技术实现思路
1、有鉴于此,本发明的一个目的是开发一类安全有效、结构新颖的4-(3-吲哚基)-1,3-噻唑类化合物及其制备方法和应用,该类化合物作为lsd1抑制剂具有较好的临床应用前景,为靶向lsd1的创新药物开发提供物质基础。
2、为此,本发明提供的技术方案为:一种4-(3-吲哚基)-1,3-噻唑类化合物,包括结构通式i所示的4-(3-吲哚基)-1,3-噻唑类化合物或其药学上可接受的盐:
3、
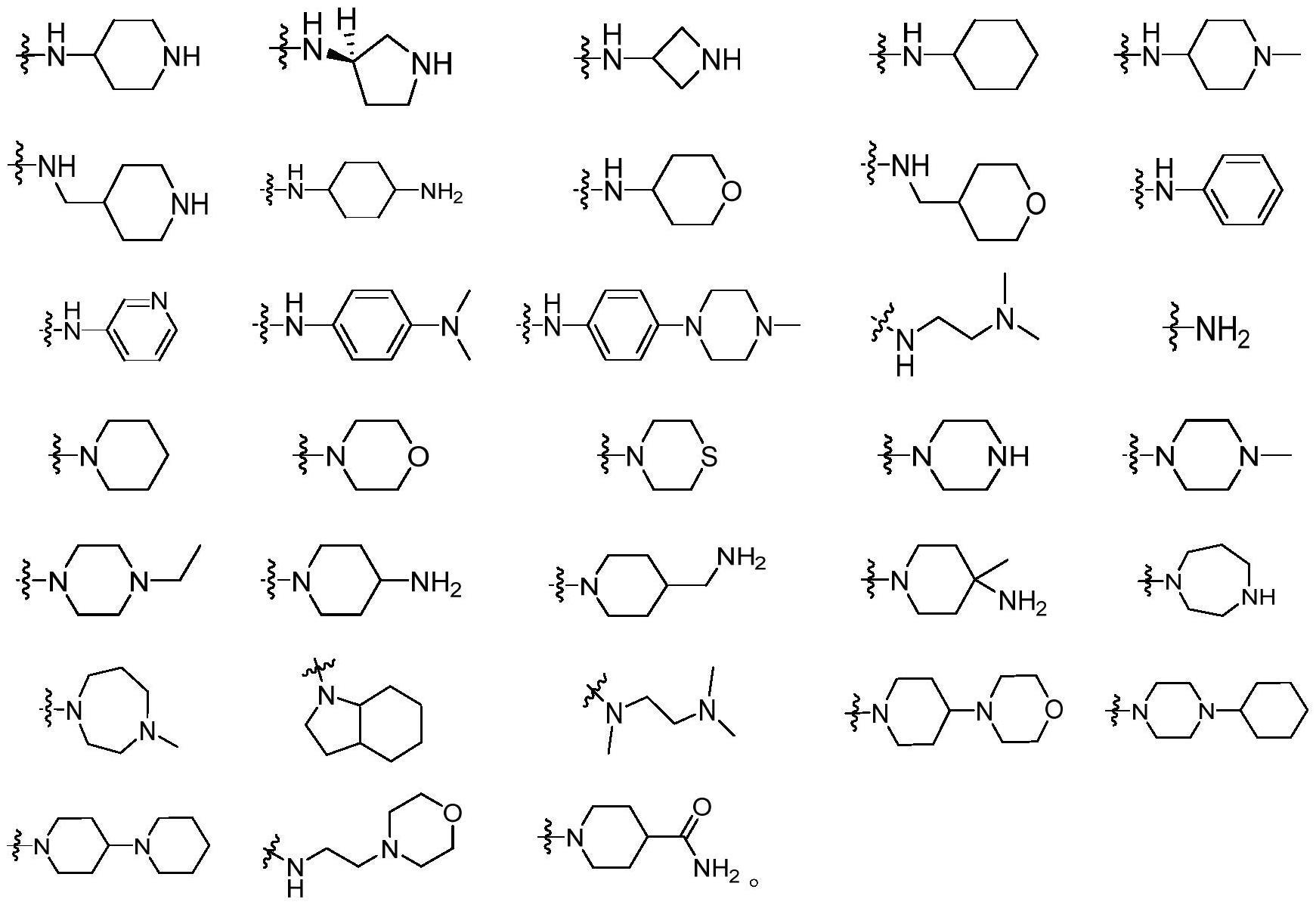
4、其中r1为氨基、c1-c10n-单取代胺基、c1-c10n,n-双取代胺基、氮杂4~8元脂肪环基、苯胺基、取代苯胺基、杂芳香胺基;“取代苯胺基”为苯环上一个或者多个h原子被二甲胺基或n-甲基哌嗪等其他基团取代,“杂芳香胺基”是苯环中至少一个碳原子被n、s或o等其他杂原子取代。r1优选以下基团:
5、
6、r2为h、c1-c8直链或支链烷基、磺酰基、甲酸酯基、苯基、取代苯基、杂芳香基、苄基、取代苄基,其中,“取代苯基”为苯环上至少有一个取代基,“取代苄基”为苄基中苯环上至少有一个取代基;“杂芳香基”是指苯环中至少一个碳原子被其他杂原子如n、s或o取代。r2优选以下基团:
7、
8、
9、r3为h、苯基、取代苯基、杂芳香基,“取代苯基”为苯环上至少有一个h原子被硝基、氰基、卤素、胺基或烷氧基等基团取代;“杂芳香基”是指苯环中至少一个碳原子被其他杂原子如n、s或o取代;r3优选以下基团:
10、
11、上述4-(3-吲哚基)-1,3-噻唑类化合物药学可接受的盐为三氟乙酸盐、硫酸盐、盐酸盐、甲磺酸盐等。
12、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
13、中间体b的制备:3-乙酰吲哚在碱性条件下进行boc保护合成中间体a,该中间体a与溴化铜发生溴代反应,合成中间体b;
14、中间体d的制备:苯甲酰基异硫氰酸酯与胺r1nh2反应,合成中间体c;中间体c在碱性条件下水解,合成中间体d;
15、目标产物f的制备:中间体d和中间体d在碱性条件下进行hantzsch噻唑关环反应合成中间体e;对中间体e进行脱去boc保护基处理,获得目标产物f。
16、中间体a、b、c、d、e、f的结构式依次为:
17、所述中间体b的制备包括:3-乙酰吲哚在碱性条件下将吲哚1位boc保护得到化合物a,将化合物a于chcl3/etoac/meoh(1/1/0.1,v/v/v)中使用溴化铜进行溴代,得到化合物b。其中,该步骤中使用的碱为氢化钠、三乙胺、n,n-二异丙基乙胺、吡啶,所使用的溶剂为四氢呋喃、氯仿、乙酸乙酯或甲醇。
18、所述中间体d的制备包括:苯甲酰基异硫氰酸酯在有机溶剂中与胺r1nh2反应得到中间体c;中间体c在碱性条件下于醇溶剂中裂解苯甲酰基得到中间体d。该步骤中采用的碱为碳酸钾、氢氧化钠、氢氧化钾,有机溶剂为水、甲醇或乙醇。
19、所述目标产物f的制备包括:中间体b和中间体d在碱性条件下,于醇性溶剂中进行hantzsch噻唑关环反应,得到中间体e;将e在强酸条件下脱去boc保护基,蒸干可得目标产物f。其中,该步骤中采用的碱为碳酸钾、碳酸氢钠、吡啶,采用的醇性溶剂为乙醇、甲醇、异丙醇、叔丁醇或正丁醇。
20、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
21、2,4-二溴噻唑在碱性条件下与胺r1nh2发生亲核取代反应,合成中间体g;中间体g与硼酸酯在催化剂作用下发生偶联反应,合成中间体h;对中间体h进行脱boc保护基处理,获得目标产物f。其中,中间体g、h以及硼酸酯的结构式依次为:
22、具体地,2,4-二溴噻唑在碱性条件下与胺发生亲核取代反应得到g,其在pdcl2(dppf)条件下与硼酸偶联得到h,h在强酸条件下脱去boc保护基得到化合物f。目标产物f在制备过程中采用的碱为碳酸钾、三乙胺、dipea,所采用的溶剂为二氯甲烷、dmf(n,n-二甲基甲酰胺)、二氧六环或水。
23、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
24、3-乙酰吲哚在碱性条件下与碘代物r2-i发生乌尔曼(ullmann)偶联反应,合成中间体i;中间体i与溴化铜发生溴代反应,合成中间体j;中间体j与4-吗啉硫代甲酰胺在碱性条件下发生hantzsch噻唑环合反应,合成目标产物k。其中,中间体i、j、k的结构式依次为:
25、具体地,3-乙酰吲哚在碱性条件下与碘代物发生ullmann偶联反应得到中间体i,中间体i于chcl3/etoac/meoh(1/1/0.1,v/v/v)中使用溴化铜进行溴代得到中间体j;在碱性条件下,中间体j与4-吗啉硫代甲酰胺于醇性溶剂中进行hantzsch噻唑环合反应得到目标产物k。目标产物k在制备过程中采用的碱为氢化钠或吡啶,采用的溶剂为dmso、氯仿、乙酸乙酯、甲醇、乙醇。
26、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
27、化合物17与r2x在碱性条件下经亲核取代反应得到目标产物m;r2x中的x为卤素。其中,目标产物m的结构式为其中,目标产物m合成过程过程中使用的碱为氢化钠、dmap,使用的溶剂为dmf、四氢呋喃或二氯甲烷。
28、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
29、化合物41和液溴发生亲电取代反应,合成中间体p;中间体p和r3-b(oh)2发生偶联反应,合成中间体r;对中间体r进行脱去boc保护基处理,获得目标产物s。其中,中间体p、r及目标产物s的结构式依次为:
30、具体地,化合物41在液溴作用下,于非极性溶剂中发生亲电取代反应得到化合物p;化合物p在pdcl2(dppf)条件下与取代硼酸r3-b(oh)2进行偶联得到化合物r,接着r强酸作用下脱去boc可得化合物s。在目标产物s制备过程中,采用的碱为碳酸氢钠、碳酸钾,采用的溶剂为1,4-二氧六环、水、二氯甲烷。
31、本发明还提供一种4-(3-吲哚基)-1,3-噻唑类化合物的制备方法,包括步骤:
32、化合物t和液溴发生亲电取代反应,合成中间体u;中间体u和和3-甲氧基苯基硼酸发生偶联反应,合成中间体v;对中间体v进行脱去boc保护基处理,获得目标产物w。中间体t、u、v及目标产物w的结构式依次为:
33、化合物t在液溴作用下,于非极性溶剂中发生亲电取代反应得到化合物u,化合物u在pdcl2(dppf)条件下与3-甲氧基苯基硼酸偶联得到化合物v,接着v强酸作用下脱去boc可得化合物w。所述过程中使用的碱可能为碳酸氢钠、碳酸钾,所使用的溶剂为1,4-二氧六环、水、二氯甲烷。
34、本发明还提供一种上述4-(3-吲哚基)-1,3-噻唑类化合物在制备lsd1抑制剂中的应用。
35、因此,经试验证明:本发明提供的上述4-(3-吲哚基)-1,3-噻唑类化合物具有抑制lsd1的作用,为寻找一类新的基于lsd1靶点的创新药物发现开辟一条新途径。
- 还没有人留言评论。精彩留言会获得点赞!