一种嘧啶胺类NUAK抑制剂及其制备方法和用途与流程
本发明属于小分子化合物领域,具体地,涉及一种嘧啶胺类nuak抑制剂及其制备方法和用途。所述化合物通过抑制nuak1/nuak2,可用于预防或治疗以下疾病:神经精神类疾病、代谢性疾病、肿瘤、内脏纤维化疾病、皮肤纤维化疾病,或单独用于减轻外伤和手术后疤痕。
背景技术:
1、蛋白激酶是一组参与调节细胞代谢、极性、生长、分裂和分化的重要功能性蛋白,人类基因组编码500多种蛋白激酶,这些激酶能将atp(adenosine triphosphate)上的磷酸基团转移到底物蛋白的特定丝氨酸、苏氨酸或酪氨酸残基上而使之磷酸化;大部分蛋白激酶为丝氨酸/苏氨酸激酶,酪氨酸激酶不到100种。单磷酸腺苷活化蛋白激酶(adenosinemonophosphate-activated protein kinase,ampk)属于丝氨酸/苏氨酸蛋白激酶(serine/threonine kinases,stk),是哺乳动物体内细胞能量稳态(cellular energyhomeostasis)的重要调节因子,通过感应代谢压力(如缺氧,热休克)下细胞内amp(adenosine monophosphate)/atp、或adp(adenosine diphosphate)/atp比例变化来调节糖和脂类代谢、细胞增生和细胞极性,可谓代谢感应蛋白(metabolic sensor protein)。ampk在进化上非常保守,是一种异三聚体蛋白,由含有激酶结构域(kinase domain,kd)的α亚单位、以及调节激酶活性的β亚单位和γ亚单位组成,γ亚单位上含有四个cbs功能域(cystathionine-β-synthase(cbs)domains),负责检测细胞内amp/atp和adp/atp比例的变化(hardie dg,trends in cell biology.2016,26:190)。
2、ampk功能紊乱会导致肥胖、糖尿病、炎症性疾病和肿瘤等多种疾病,因此机体对ampk功能的调控非常关键。首先ampk的激活需要上游激酶对其α亚单位第172位上的苏氨酸(t172)进行磷酸化,目前已知的ampk上游激酶有三种:肝激酶b1(liver kinase b1,lkb1),钙离子/钙调蛋白依赖性蛋白激酶2(ca2+/calmodulin-dependent pk kinase 2,camkk2),以及转化生长因子β激活激酶1(transforming growth factor-β-activated kinase 1,tak1)。与上游激酶通过磷酸化t172位点激活ampk相对应,已知有三种蛋白磷酸酶能通过移除t172上的磷酸基来抑制ampk的活性,包括蛋白磷酸酶2a(protein phosphatase 2a,pp2a),蛋白磷酸酶2c(protein phosphatase 2c,pp2c),以及镁/锰依赖性蛋白磷酸酶1e(mg2+-/mn2+-dependent protein phosphatase 1e,ppm1e)。当细胞处于低能状态时(高amp/atp或adp/atp比例),ampk的α亚单位kd和γ亚单位紧密交联,同时β亚单位豆蔻酰化,确保蛋白磷酸酶无法接触到t172位点而保持激活状态。而当细胞处于高能状态时,kd和γ亚单位松开,t172暴露给磷酸酶而导致ampk失活(steinberg gr,nature reviews drugdiscovery.2019,18:527)。
3、除去ampk上游激酶和蛋白磷酸酶能分别激活和灭活ampk,还有一类ampk相关性激酶(ampk-related kinases,arks)参与调节ampk的功能,目前一共发现有12种arks(brsk1,brsk2,nuak1,nuak2,qik,qsk,sik,mark1,mark2,mark3,mark4和melk),都属于丝氨酸/苏氨酸蛋白激酶,arks的激酶结构域和ampk的α亚单位有很高的同源性,除melk外都能被lkb1激活,并且其激酶活化的磷酸化位点也和ampk的t172相当,功能上也都参与细胞代谢、增生和极性的调控。不过由于不像ampk具备调控亚单位,arks不能直接被细胞内amp/atp比例所调控(bright nj,acta physiologica.2009,196:15)。
4、arks又因蛋白结构和功能特征的不同而分成数个ark亚家族,其中nuak(nu(novel)and ampk-related kinase)ark亚家族含有nuak1(最早被命名为ark5)和nuak2(最早被命名为sucrose nonfermenting-like/ampk related kinase,snark)两个成员,两者的氨基酸序列大约55%同源,根据氨基酸序列,nuak1和nuak2的推测分子量分别为76和69kda;其蛋白结构很相近,氨基端为激酶结构域,羧基端为泛素交联域(ubiquitin-associated domain),目前尚不清楚nuak是否和ampks一样也是异三聚体结构。nuak1和nuak2在大部分组织中有表达,其中nuak1在脑、皮肤、肌肉、消化道上段、内分泌等器官组织中表达明显高于其它部位,nuak2表达最高的器官组织为消化道、女性生殖系统、皮肤、骨骼、脑和内分泌等,其中nuak2的表达更多地显示出组织特异性。值得一提的是,nuak1和其它arks及ampk蛋白主要分布在细胞浆内,而nuak2则主要分布在细胞核内,并有迹象表明nuak2作为转录调节因子参与代谢压力相关的基因表达(sun x,j of molecularendocrinology.2013,51:r15)。
5、作为丝氨酸/苏氨酸蛋白激酶,nuak1和nuak2可磷酸化多种蛋白底物,包括参与细胞信号传导、代谢、细胞增殖、细胞凋亡、自噬和细胞骨架组织的蛋白质。已知nuak1可以磷酸化ampk,lats1/2,p53肿瘤抑制蛋白,以及调节肌动蛋白细胞骨架组织的肌球蛋白磷酸酶靶亚基1(mypt1)等。nuka2能磷酸化hedghog信号通路的转录因子gli3,foxo1,驱动蛋白轻链1(klc1),以及rho gdp解离抑制剂α(rho gdiα)等。
6、nuak1和nuak2的功能调控涉及多种细胞内信号系统,除了能被lkb1磷酸化激活(t211,相当于ampk的t172)和被钙离子/pkc活化,nuak1是ampk相关蛋白家族里唯一能被akt激活的成员,nuak1活性升高也见于一些生长因子如胰岛素样生长因子1(insulin-likegrowth factor 1,igf1)信号通路的激活,骨骼肌细胞处于收缩状态时也伴随nuak1的活性增强。nuak1和nuak2都能被lkb1激活(t211/nuak1,t208/nuak2,相当于ampk的t172),但是nuak2可以进行自磷酸化活化,nuak2能与x染色体上的泛素特异性蛋白酶9(ubiquitin-specific protease 9,x chromosome,usp9x)进行交互作用,后者是一种去泛素化酶,能够维持nuak2的活性。在不同的细胞内,低渗透压、dna损伤、氧化、营养不足等刺激均可导致nuak2的激活,nuak2的活化也见于骨骼肌细胞细胞的收缩(brooks d,data inbrief.2022,43:108482)。
7、多项研究表明nuak在代谢性疾病、肿瘤、神经退行性疾病和纤维化疾病等疾病的发病过程中扮演重要的角色,nuak1和nuak2纯合敲除基因鼠基本都导致胚胎期死亡,nuak1杂合缺失小鼠导致机体和神经组织(如脑皮质和外周神经元)发育不良,人nuak1杂合基因突变与孤独症(autism spectrum disorders,asd),认知缺陷,多动症(attentiondeficit/hyperactivity disorder,ad/hd)及精神分裂症(schizophrenia)相关。人nuak2基因缺失导致无脑儿的形成,是一种引起胎儿发育缺陷的严重神经管畸形,其机理和yap的功能丧失有关(bonnard c,j exp med.2020,217:e20191561)。骨骼肌细胞特异性敲除nuak1基因可以避免高脂饮食诱导的亚临床糖尿病;而nuak2杂合缺失小鼠出现的表型则与人二型糖尿病伴肥胖的一系列临床表现相近,这些现象可能与细胞自噬机制失衡有关(bennison sa,cellular signaling.2022,100:110472;blazejewski sm,scientificreports.2011,11:8156)。
8、akt和其它蛋白激酶导致的nuak1激活能促进肿瘤细胞在能量不足的环境下生存并保护肿瘤细胞免于发生凋亡,nuak2也通过类似的机制抑制tnfα和cd95诱导的细胞凋亡。此外,nuak1激活后通过上调机制金属蛋白酶(matrix metalloproteinases,mmp)促进肿瘤细胞的侵袭和转移,nuak2则通过加强肿瘤细胞的活动性参与肿瘤转移的发生(hou x,oncogen.201,30:2933;chen y,cell death and disease.2020,11:712;molina e,cells.10:2760;humbert n,the embo journal.2010,29:376)。很多证据表明nuak1和nuak2在肿瘤形成和转移中发挥作用,比起nuak1,nuak2存在更强的促进肿瘤形成和转移的作用,nuak2抑制剂应该比nuak1抑制剂更有可能成为新的靶向抗肿瘤药物。
9、最新研究表明nuak通过和转化生长因子-β(tgf-β)信号通路交互影响在组织纤维化的发展中起着关键作用。首先tgf-β能在多种上皮细胞如角质形成细胞(keratinocytes)和人皮肤纤维母细胞(dermal fibroblasts)内上调nuak1和nuak2的基因转录,而mapk(erk1/2and p38)信号失活抑制tgf-β依赖性nuak2表达;此外,nuak2通过和tgf-β的细胞内信号中介smad3蛋白结构上的交联域和mh2域结合来抑制smad3在细胞内的降解,类似的机制在nuak2和tβri之间也存在。tgf-β诱导的编码纤连蛋白(fibronectin,fn),纤溶酶原激活物抑制剂1(plasminogen activator inhibitor 1,pai1),组织金属蛋白酶抑制蛋白1(tissue inhibitor of metalloproteinase-1,timp1)等促纤维化分子的基因表达依赖于nuak2的存在,可见nuak2促进纤维化的发生。研究也表明nuak1通过上调tgf-β和yap两个信号通路促进肾、肺和肝纤维化,动物试验中抑制nuak1能减轻新创伤引起的瘢痕和陈旧性瘢痕组织(gill mk,nature communications.2018.9:3510)。有趣的是,研究发现nuak1敲基因的角质形成细胞内编码fn的基因表达上调,nuak1可能通过负反馈机制影响tgf-β信号传导从而抑制纤维化(van de vis raj,cancers.2021,13:3377)。总体来讲,抑制nuak可能可以有效抑制参与组织器官的纤维化进程。
10、由于激酶从结构和功能上一直是理想的小分子药物靶点,而nuak1和nuak2又参与多种疾病的发生,目前已经有一些nuak抑制剂处于早期研发阶段,其有效性和安全性依然存疑(banerjee s,the biochemical journal.2014,457:215)。因此开发nuak选择性抑制剂和nuak1/2双靶点抑制剂有望为神经精神类疾病(如帕金森氏病,阿尔茨海默氏病)、代谢性疾病(如糖尿病,高脂血症,肥胖)、肿瘤(如肝癌,白血病,淋巴瘤,肿瘤转移)、内脏纤维化疾病(如肝硬化,肾纤维化,肺纤维化,和心肌炎后遗症)以及皮肤纤维化疾病(如硬皮病,瘢痕疙瘩,肥厚性瘢痕,或单纯用于减轻外伤和手术后瘢痕)等提供创新治疗的新方向,开发新一代nuak抑制剂具有巨大的潜在临床应用价值。
技术实现思路
1、本发明的目的旨在获得有效的nuak1/nuak2抑制剂,可用于制备预防或治疗以下疾病的药物:神经精神类疾病(如帕金森氏病,阿尔茨海默病)、代谢性疾病(如糖尿病,高脂血症,肥胖)、肿瘤(如肝癌,白血病,淋巴瘤,肿瘤转移)、内脏纤维化疾病(如肝硬化,肾纤维化,肺纤维化,和心肌炎后遗症)以及皮肤纤维化疾病(如硬皮病,瘢痕疙瘩,肥厚性瘢痕,或单纯用于减轻外伤和手术后瘢痕)。
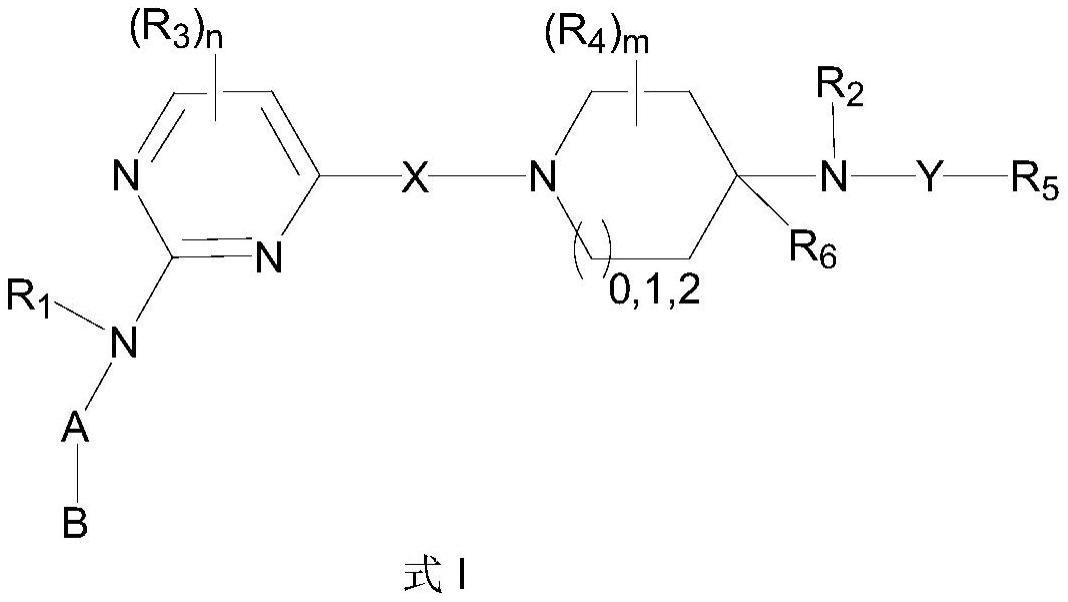
2、为了实现上述目的,在一方面,本发明提供了一种嘧啶胺类化合物,所述嘧啶胺类化合物为由如下所示的式i表示的化合物,或其立体异构体、几何异构体、互变异构体、同位素衍生物、水合物、溶剂化物、前药以及药学上可接受的盐:
3、
4、其中,x为直接键、-o-、-s-、-nr7-或-cr8r9-,y为-co-或-s(o)2-;
5、a为任选取代的6-10元芳基或5-10元杂芳基,b为任选取代的3-10元环烷基或3-10元杂环烷基,a或b上的取代基选自卤素、氨基、羟基、硝基、氰基、巯基、任选取代的c1-8烷基、任选取代的c1-8烷基氧基、任选取代的c1-8烷基硫基、任选取代的c1-8烷基氨基的一个或多个;
6、r1、r2、r6、r7、r8、r9各自独立地为h、任选取代的c1-8烷基;
7、r3、r4各自独立地为卤素、氨基、羟基、硝基、氰基、巯基、任选取代的c1-8烷基、任选取代的c1-8烷基氧基、任选取代的c1-8烷基硫基、任选取代的c1-8烷基氨基;
8、r5为任选取代的3-10元环烷基、任选取代的3-10元杂环烷基,其中环烷基或杂环烷基上的取代基选自卤素、氨基、羟基、硝基、氰基、巯基、任选取代的c1-8烷基、任选取代的c1-8烷基氧基、任选取代的c1-8烷基硫基、任选取代的c1-8烷基氨基中的一个或多个;
9、c1-8烷基上的取代基选自卤素、氨基、羟基、硝基、氰基、巯基中的一个或多个;
10、n为0、1或2,m为0、1、2、3、4或5。
11、在一组实施方式中,r5为任选取代的3-10元环烷基,优选任选取代的3-6元环烷基,优选为卤素取代的3-6元环烷基,更优选r5为氟取代的3-6元环烷基。
12、进一步地,r5为优选r5为
13、在一组实施方式中,r6为甲基。
14、在一组实施方式中,r2为h。
15、在一组实施方式中,b为任选取代的3-6元杂环烷基,优选b为任选取代的5-6元杂环烷基,更优选b为任选取代的哌啶基或任选取代的哌嗪基。
16、在一组实施方式中,所述式i表示的化合物为式i-1或式i-2,
17、
18、r10选自卤素、氨基、羟基、硝基、氰基、巯基、任选取代的c1-8烷基、任选取代的c1-8烷基氧基、任选取代的c1-8烷基硫基、任选取代的c1-8烷基氨基;
19、r11为h或任选取代的c1-8烷基;
20、c1-8烷基上的取代基选自卤素、氨基、羟基、硝基、氰基、巯基中的一个或多个;
21、q为0、1、2、3或4。
22、进一步地,r11为h或甲基。
23、在一组实施方式中,a为任选取代的苯基或任选取代的5元杂芳基。
24、进一步地,a为任选取代的以下基团之一:
25、
26、
27、优选上方与氨基相连,下方与b基团相连。
28、在一组实施方式中,r3为氯,优选取代在氨基的对位。
29、在一组实施方式中,a环上的取代基为甲氧基,优选取代在氨基的邻位。在一组实施方式中,所述式i表示的化合物为以下化合物之一:
30、
31、
32、在一组实施方式中,所述药学上可接受的盐为甲酸盐。
33、在一组实施方式中,所述同位素衍生物为氘代物。
34、另一方面,本技术还提供了所述嘧啶胺类化合物的制备方法,其包含以下步骤:由式ii化合物和式iii化合物制备式i化合物;
35、或
36、由式iv与式ii化合物制备式v化合物,再由式v化合物制备式i化合物;
37、任选地,制备过程中包含必要的保护和脱保护步骤;
38、
39、
40、其中,z为离去基团,优选z为卤素,更优选z为cl,其他各基团的定义如上所定义。
41、在一组实施方式中,先对式ii中b环上的氨基或亚氨基进行保护,与式iii化合物偶联后,再脱除保护基制备式i化合物。
42、在一组实施方式中,先对式iv化合物中r2所连氨基或亚氨基进行保护,任选对式ii中b环上的氨基或亚氨基进行保护,之后由经保护的式iv化合物与任选保护的式ii化合物偶联、脱除r2所连氨基或亚氨基上的保护基制备式v化合物,再由式v化合物与r5-y-oh反应、任选脱除b环上的保护基制备式i化合物。
43、本技术提供了一种药物组合物,以本技术以上所述的嘧啶胺类化合物作为有效成分;药物组合物进一步包含药学上可接受的载体或赋形剂。
44、本技术提供了所述嘧啶胺类化合物用于制备nuak1或nuak2抑制剂的用途。
45、本技术提供了所述嘧啶胺类化合物用于制备药物的用途,其特征在于所述化合物通过抑制nuak1或nuak2用于预防或治疗以下疾病:神经精神类疾病、代谢性疾病、肿瘤、内脏纤维化疾病、皮肤纤维化疾病。
46、在一组实施方式中,所述疾病为帕金森氏病、阿尔茨海默病、糖尿病、高脂血症、肥胖、肝癌、白血病、淋巴瘤、肿瘤转移、肝硬化、肾纤维化、肺纤维化、心肌炎后遗症、硬皮病、瘢痕疙瘩、肥厚性瘢痕、或单独用于减轻外伤和手术后疤痕。
47、本发明的有益效果为:
48、本发明提供了一类嘧啶胺类化合物,体外激酶活性抑制试验显示,本发明化合物对nuak1/nuak2激酶具有优异的抑制活性。可通过抑制nuak1或nuak2用于预防或治疗以下疾病:神经精神类疾病、代谢性疾病、肿瘤、内脏纤维化疾病、皮肤纤维化疾病。
- 还没有人留言评论。精彩留言会获得点赞!