一种靶向AR-V7的具有约束的螺旋和片状结构的双订书肽的制备方法和应用
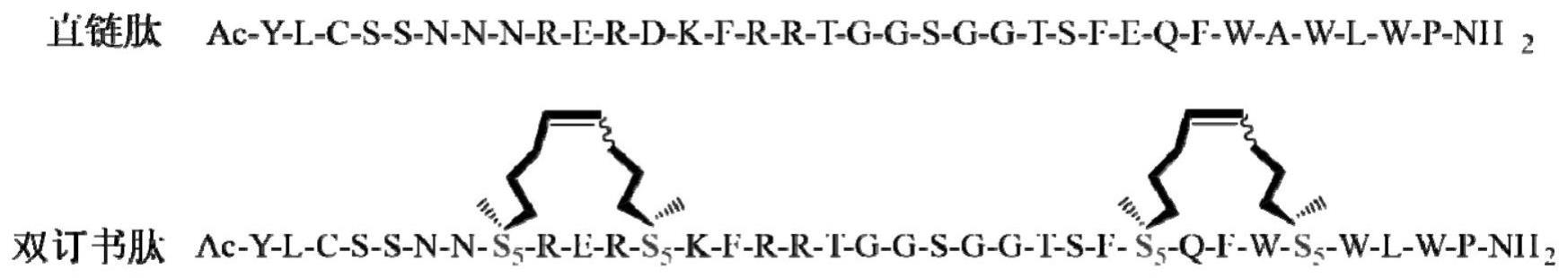
本发明涉及多肽药物领域,具体地说,是一种靶向ar-v7的具有约束的螺旋和片状结构的双订书肽的制备方法和应用。
背景技术:
1、靶向蛋白降解(targeted protein degradation,tpd)是近年来备受关注的一种新兴治疗策略。tpd试剂的开发涉及被称为“降解剂”或“蛋白水解靶向嵌合体”(protacs)的分子的设计和合成,其由两个功能部分组成:与靶蛋白结合的配体和与e3泛素连接酶结合的配体。e3连接酶负责催化泛素分子附着在靶蛋白上,导致其被蛋白酶体降解。目前,基于小分子的protac药物开发领域已经取得了实质性进展,以arv-110和arv-471为代表的许多化合物已经进入临床试验阶段。然而,当涉及到不具有明确可识别的口袋结合转录因子(包括ar-v7)的靶标时,小分子protacs的进展缓慢,基于肽的protac药物已成为一种有前途的替代解决方案。
2、然而,由于前几代多肽药物的血液稳定性差和细胞膜渗透能力有限等问题,其发展面临挑战。多肽药物的递送系统和化学修饰的最新进展开辟了其在protac药物临床应用中的可能性。订书修饰肽作为一种增强多肽稳定性和提高其细胞穿透能力的策略备受关注。
技术实现思路
1、本发明的目的在于提供一种靶向ar-v7的具有约束的螺旋和片状结构的双订书肽的制备方法和应用。
2、为了提高靶向ar-v7的肽类protac药物的临床潜力,本发明采用双订书肽修饰策略,本发明的双订书肽修饰方法旨在在肽序列内产生稳定的α螺旋或β片结构,或两者兼有。这种结构稳定性增强了多肽与目标蛋白的结合亲和力。这些修饰已被证明能有效增强多肽药物的药代动力学和药效学特性,确实与线性肽相比,表现出显著改善的蛋白水解稳定性和增强的生物活性。从而为提高ar-v7的靶向多肽protac药物的临床应用提供了巨大的潜力。
3、为了实现上述目的,本发明采用以下技术方案:
4、本发明设计了一种名为dsartc的双订书肽药物,专门针对ar dna结合域。研究结果揭示了dsartc在降解ar和ar-v7以及抑制前列腺癌细胞增殖方面的巨大潜力。并且表明双订书修饰方法,可以成为开发肽protac药物的可行策略。这些发现强调了在多肽中使用多个短链修饰的潜在益处,从而增强了多肽药物的可药性和治疗潜力。
5、本发明的第一方面,提供一种订书肽,所述的订书肽为:
6、以ac-ylcssnnnrerdkfrrtggsggtsfeqfwawlwp-nh2(seq id no.1)为肽链模板,其中8n、12d、26e和30a被s5替换并环合。
7、进一步的,所述的订书肽的结构示意图如图1所示;其结构式如下所示:
8、
9、本发明的第二方面,提供一种如上所述的订书肽的制备方法,包括以下步骤:
10、(a)在缩合试剂的作用下分别使c端首个氨基酸的cooh与固相载体氨基树脂偶联;
11、(b)使用脱保护试剂脱去氨基酸上的fmoc保护基;
12、(c)在缩合试剂作用下连接下一个氨基酸;
13、(d)重复进行脱保护-耦合操作,按照从c端至n端的顺序,依照氨基酸序列合成肽链;其中,环合位点以s5分别替代i和i+4位氨基酸;其中8n、12d为一组i和i+4,26e和a30为一组i和i+4;
14、(e)最后一个氨基酸脱保护后乙酰化;
15、(f)在环合试剂作用下使i和i+4位s5氨基酸发生烯烃复分解反应,环合肽链;
16、(g)使用切割试剂将肽链从载体上切下,纯化后得相应订书肽。
17、进一步地,步骤(a)中采用的缩合试剂为dic-oxyme缩合体系,以nmp为溶剂。
18、更进一步地,步骤(a)中氨基酸、oxyme、dic、nmp的比例为1:1:1:6(mol/mol/mol/ml)或1:0.9:0.9:6(mol/mol/mol/ml)。
19、进一步地,步骤(a)中固相合成时,树脂的载样量为0.30mmol/g。
20、进一步地,步骤(a)中偶联反应的温度为60℃;偶联反应的时间为20min。
21、进一步地,步骤(b)中,所述脱保护试剂为oxyme、哌啶及dmf的混合溶液,比例为71:2:4(m/v/v)。
22、进一步地,步骤(b)中,脱fmoc保护是采用保护试剂作用5min,两遍;脱除fmoc基团的反应温度为20~30℃,更优选为25℃。
23、进一步地,s5和s5后所接的第一个氨基酸反应时间为2h并按相同条件重复反应一次再进行下一步操作。
24、进一步地,步骤(e)中,使用的乙酰化试剂为diea、dmf与醋酸酐的混合液,投料比为1:1:8(v/v/v)。
25、进一步地,步骤(e)所述乙酰化是采用树脂在乙酰化试剂中反应20min;反应温度为20~30℃,更优选为25℃。
26、进一步地,步骤(f)中所述环合试剂为grubbsⅰ试剂的二氯乙烷的溶液,投料比为树脂量:grubbsⅰ试剂:二氯乙烷=0.3:58:6(mmol/mg/ml)。
27、进一步地,步骤(f)中所述环合是树脂在环合试剂中震荡两次,每次2h;反应温度为20~30℃,更优选为25℃。
28、进一步地,步骤(g)中,所述切割试剂为tips、tfa、h2o和苯酚的混合溶液,体积比为2:88:5:5,所述切割试剂与直链肽的体积质量比为1:10(ml/mg)。
29、进一步地,步骤(g)中,切割的温度为20~30℃,更优选为25℃;切割的时间为4h。
30、进一步地,步骤(g)采用的纯化方法为反向高效液相色谱法,条件如下:色谱柱:ymc-pack ods-aq柱;流动相:流动相a为体积分数为0.1%tfa/水,流动相b为体积分数为0.1%tfa/乙腈;梯度洗脱程序:35%b洗脱0~5min,35%b~55%b、5~60min;流速为5ml/min,进样量为1ml,检测波长214nm和254nm。
31、本发明的第三方面,提供一种如上所述的订书肽在制备抗前列腺癌药物中的应用。
32、本发明中涉及的缩略词解释如下:
33、fmoc:芴甲氧羰基
34、dcm:二氯甲烷
35、dce:二氯乙烷
36、dmf:n,n-二甲基甲酰胺
37、oxyme:ethyl cyanoglyoxylate-2-oxime,2-肟氰乙酸乙酯
38、dic:n,n-二异丙基碳二亚胺
39、nmp:n-甲基吡咯烷酮
40、diea:n,n-二异丙基乙胺
41、s5:2-amino-2-methylhept-6-enoic acid,氨基-2-甲基-6-庚烯酸
42、tfa:三氟乙酸
43、tips:三异丙基硅烷
44、grubbsⅰ:苯基亚甲基双(三环已基磷)二氯化钌
45、本发明优点在于:
46、1、本发明以氨基树脂为固相载体,以直连肽ac-ylcssnnnrerdkfrrtggsggtsfeqfwawlwp-nh2为模板,在dic-oxime缩合体系中,通过fmoc固相合成法,合成得到肽链,其间在保留关键氨基酸残基的基础上,于特定位置以s5代替原有氨基酸,直链肽在grubbsⅰ试剂的二氯乙烷溶液中进行烯烃复分解反应环合后从树脂上切下得到订书肽,所得化合物经纯化后,采用hplc及ms等进行分析表征。该方法简便易行,所得订书肽纯度大于98%,产率较高。
47、2、本发明采用双订书肽修饰策略,旨在在多肽序列内产生稳定的α螺旋或β片结构,或两者兼有。这种结构稳定性增强了多肽与目标蛋白的结合亲和力。能有效增强多肽药物的药代动力学和药效学特性,确实与线性肽相比,表现出显著改善的蛋白水解稳定性和增强的生物活性。
48、3、本发明的双订书肽可以显著抑制前列腺癌细胞的生长繁殖,在治疗前列腺癌疾病方面具有潜在的应用价值。本发明突出了双订书肽protac药物作为一种新的有前途的治疗方法的潜力,并为开发针对具有挑战性的疾病靶点的靶向治疗开辟了新的可能性。
- 还没有人留言评论。精彩留言会获得点赞!