咪唑并噻唑修饰的萘酰亚胺-多胺缀合物、制备方法和用途
本发明属于药物合成,具体涉及一种咪唑并噻唑修饰的萘酰亚胺-多胺缀合物、制备方法和用途。
背景技术:
1、咪唑并噻唑分子中包含咪唑环和噻唑环两个共轭杂环,从1987年andreani发现咪唑并噻唑衍生物具有利尿活性后,该类化合物逐渐受到化学家、药学家们的广泛关注。研究发现通过在噻唑环或咪唑环引入不同取代基,可使咪唑并噻唑衍生物具有抗菌、免疫调节、抗病毒、杀虫、抗心律失常和强心等生物活性。如作为广谱驱肠虫药和生物反应调解药物的左旋咪唑,其主要结构单元就是咪唑并噻唑。针对咪唑并噻唑,目前的研究热点是通过在咪唑和噻唑环中引入各种功能性基团,来提高该类衍生物的药理活性及扩大其药理活性范围。同时,长期实践证实,取代基团及取代位置的不同会不同程度的影响衍生物的活性。
2、萘酰亚胺类化合物具有广泛的生物活性,一直是药物化学家们研究的热点之一。萘酰亚胺-多胺缀合物在保持萘酰亚胺类化合物较好体内外抗肿瘤活性的同时,因多胺片段的引入,赋予了该类衍生物特殊的生理活性,如萘酰亚胺-多胺缀合物能靶向定位到细胞亚器线粒体、溶酶体等,在发挥抗肿瘤作用的同时可作为活细胞荧光探针,可用来开发抗肿瘤双功能试剂。
3、然而,目前现有技术并未有将咪唑并噻唑基团与萘酰亚胺、多胺进行共修饰以发挥抗肿瘤作用的相关研究。因此,如何利用咪唑并噻唑对萘酰亚胺、多胺化合物进行有效的结构修饰以提高化合物的抗肿瘤活性并实现新型抗肿瘤药物的研发,成为亟需解决的技术问题。
技术实现思路
1、本发明的第一目的在于提供一种咪唑并噻唑修饰的萘酰亚胺-多胺缀合物,其采用咪唑并噻唑修饰萘酰亚胺萘环,并与多胺缀合,形成新的药效团,该类化合物在保持萘酰亚胺类化合物活性的同时,也兼具了咪唑并噻唑类化合物的特性,能够提高目标分子的抗肿瘤活性。
2、本发明的第二目的在于提供一种咪唑并噻唑修饰的萘酰亚胺-多胺缀合物的制备方法,其工艺条件温和,便于实现萘酰亚胺化合物的结构修饰。
3、本发明的第三目的在于提供一种咪唑并噻唑修饰的萘酰亚胺-多胺缀合物在制备抗肿瘤药物中的用途。
4、本发明的目的之一采用如下技术方案实现:
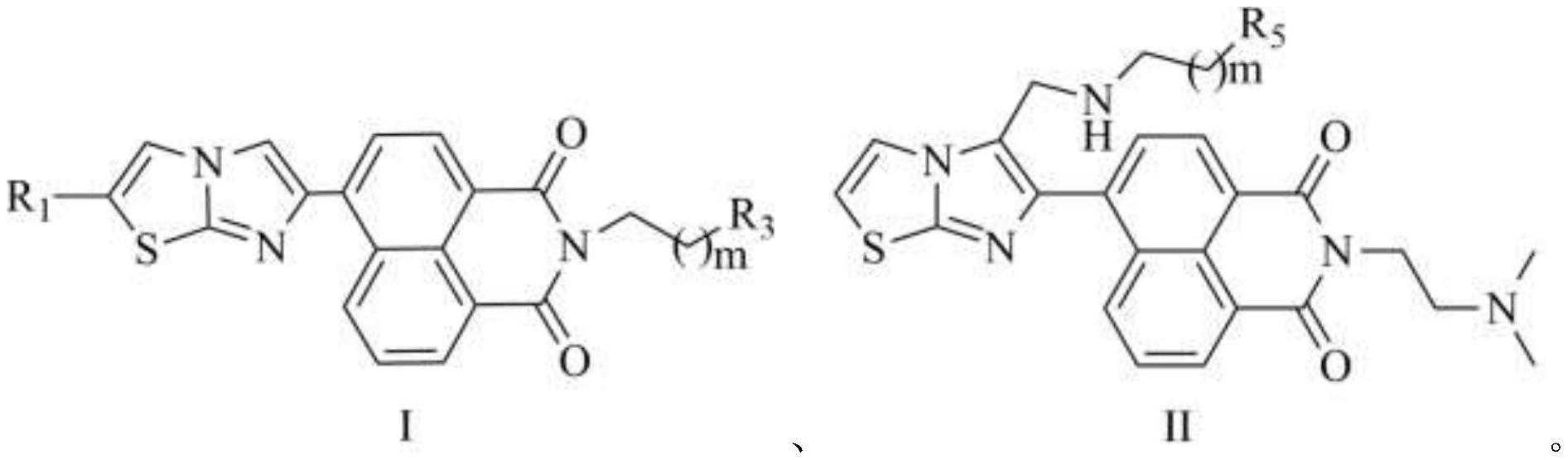
5、一种咪唑并噻唑修饰的萘酰亚胺-多胺缀合物,为通式ⅰ或通式ⅱ所示的化合物,或者通式ⅰ或通式ⅱ所示的化合物药学上可接受的盐:
6、
7、其中,通式ⅰ和通式ⅱ中,m为1、2或3;
8、通式ⅰ中,r1为氢或甲基;
9、r3选自
10、中的任一种;
11、通式ⅱ中,r5选自中的任一种。
12、本发明提供的咪唑并噻唑修饰的萘酰亚胺-多胺缀合物,采用咪唑并噻唑基团修饰萘酰亚胺萘环的6位,合成咪唑并噻唑修饰的萘酰亚胺新药效团,并与多胺缀合,设计合成了一类咪唑并噻唑修饰的萘酰亚胺-多胺缀合物。该类化合物在保持萘酰亚胺类化合物的活性的同时,也兼具了咪唑并噻唑类化合物的特性,改善了原有分子的生物学活性,提高了目标分子的抗肿瘤活性。
13、优选地,所述药学上可接受的盐为盐酸盐、硫酸盐、马来酸盐、磷酸盐、枸橼酸盐、氢溴酸盐、醋酸盐、苯磺酸盐、酒石酸盐、碳酸盐、柠檬酸盐、苹果酸盐、甲磺酸盐、硬脂酸盐、戊酸盐、硝酸盐中的一种或多种。本发明对于药学上可接受的盐的种类不做特别限定,本领域技术人员可以根据实际需求进行选择。
14、本发明的目的之二采用如下技术方案实现:
15、如上所述的咪唑并噻唑修饰的萘酰亚胺-多胺缀合物的制备方法,目标产物为通式ⅰ所示的化合物药学上可接受的盐时,制备方法包括如下步骤:
16、步骤(1):将化合物与乙酰氯反应,得到化合物
17、步骤(2):将化合物与重铬酸钠反应,得到化合物
18、步骤(3):将化合物与n-溴代琥珀酰亚胺反应,然后纯化,得到化合物
19、步骤(4):将化合物与氨基噻唑在无水乙醇中于80~90℃反应2.5~3.5h,得到化合物
20、步骤(5):将化合物与胺链r2nh2反应,纯化后得到化合物
21、步骤(6):将化合物与酸混合进行搅拌反应,得到通式ⅰ所示的化合物药学上可接受的盐;
22、目标产物为通式ⅰ所示的化合物时,制备方法除包括所述步骤(1)~步骤(6)外,还包括将步骤(6)所得通式ⅰ所示的化合物药学上可接受的盐进行中和反应以得到通式ⅰ所示的化合物的步骤;
23、目标产物为通式ⅱ所示的化合物药学上可接受的盐时,制备方法包括如下步骤:
24、步骤(a):将化合物与三氯氧磷反应,得到化合物
25、步骤(b):将化合物与胺链r4nh2反应,得到席夫碱中间体,然后采用硼氢化钠还原席夫碱中间体,得到化合物
26、步骤(c):将化合物与二碳酸二叔丁酯反应,得到化合物
27、步骤(d):将化合物与酸混合进行搅拌反应,过滤即得通式ⅱ所示的化合物药学上可接受的盐;
28、目标产物为通式ⅱ所示的化合物时,制备方法除包括所述步骤(a)~步骤(d)外,还包括将步骤(d)所得通式ⅱ所示的化合物药学上可接受的盐进行中和反应以得到通式ⅱ所示的化合物的步骤;
29、其中,所述氨基噻唑为2-氨基噻唑或2-氨基-4-甲基噻唑;
30、所述胺链r2nh2选自
31、中的一种;
32、所述胺链r4nh2选自中的一种。
33、本发明的咪唑并噻唑修饰的萘酰亚胺-多胺缀合物的制备方法,工艺条件温和,能够有效实现萘酰亚胺化合物的结构修饰。其中,上述制备方法中,步骤(4)合成咪唑并噻唑修饰的1,8-萘酐的常规反应方法是:以溴代乙酰化萘酐与氨基噻唑在乙腈做溶剂,碳酸氢钾做缚酸剂,80℃的条件下进行反应。然而,发明人经过多次试验验证后发现,上述常规条件下目标化合物产率较低,而通过不断提高反应温度和延长反应时间,收率一直未能达到30%,根本无法满足咪唑并噻唑修饰的1,8-萘酐的合成需求。基于此,本发明的发明人在多项技术调研以及反复的探索试验的基础上,改用无水乙醇做溶剂,不采用缚酸剂,将反应温度调整为80~90℃,反应时间控制在2.5~3.5h,能够将反应后咪唑并噻唑修饰的1,8-萘酐的收率提高到至70%左右,同时产物以沉淀的方式析出,通过减压抽滤能够直接得到产物,不需分离可直接用于下步反应。而本发明的该项改进,极大地简化了反应条件,使后处理变得简单,更适用于工业化生产。
34、基于保证反应物的溶解以及反应效率、产物得率的考虑,优选地,步骤(2)中,化合物与重铬酸钠的反应在冰醋酸中进行;步骤(3)中,化合物与n-溴代琥珀酰亚胺的反应在乙腈中进行;步骤(4)中,化合物与氨基噻唑的反应过程不添加缚酸剂,反应的温度为85℃,时间为3h;步骤(5)中,化合物与胺链r2nh2的反应在无水乙醇中进行;步骤(a)中,化合物与三氯氧磷的反应在n,n-二甲基甲酰胺中进行;步骤(b)中,与胺链r4nh2的反应在乙醇中进行;步骤(c)中,化合物与二碳酸二叔丁酯的反应在甲醇中进行。
35、进一步地,步骤(2)中,化合物与重铬酸钠的反应摩尔比为2∶5;步骤(3)中,化合物与n-溴代琥珀酰亚胺的反应摩尔比为2∶1;步骤(4)中,化合物与氨基噻唑的反应摩尔比为1∶1。
36、优选地,步骤(6)和步骤(d)中,所述酸为盐酸、硫酸、马来酸、磷酸、枸橼酸、氢溴酸、醋酸、苯磺酸、酒石酸、碳酸、柠檬酸、苹果酸、甲磺酸、硬脂酸、戊酸、硝酸中的一种或多种。
37、更优选地,步骤(6)和步骤(d)中,所述酸为浓度为4mol/l的盐酸-乙醇溶液。
38、本发明的目的之三采用如下技术方案实现:
39、如上所述的咪唑并噻唑修饰的萘酰亚胺-多胺缀合物在制备抗肿瘤药物中的用途。
40、优选地,所述抗肿瘤药物为抑制肿瘤细胞活性的药物。
41、进一步地,所述肿瘤细胞为sun739细胞、mcf-7细胞、pc-12细胞、hepg2细胞中的一种或多种。
42、本发明通过肿瘤细胞的抑制试验证实,具有通式ⅰ或通式ⅱ所示结构的咪唑并噻唑修饰的萘酰亚胺-多胺缀合物,对sun739(人肝癌细胞)、mcf-7(人乳腺癌细胞)、pc-12(大鼠肾上腺嗜铬细胞瘤细胞)和hepg2(人肝癌细胞)多种肿瘤细胞增殖均显示出明显的抑制活性,为新型的抗肿瘤药物以及抗肿瘤先导化合物的开发提供了新方向。
- 还没有人留言评论。精彩留言会获得点赞!