订书肽及其制备方法和应用
本发明属于多肽药物领域,具体涉及一种可以在模板多肽的基础上提高抗革兰氏阳性和革兰氏阴性菌抗菌活性的订书肽及其制备方法和应用。
背景技术:
1、抗生素滥用导致耐药菌株的出现,由耐药菌株引起的感染疾病已成为全球公共卫生的严重威胁。临床上的耐药菌株,如金黄色葡萄球菌,铜绿假单胞菌,鲍曼不动杆菌等能在自身表面形成一层生物被膜,生物膜包裹的细菌对常规抗生素的耐受性是其他细菌的10~1000倍,致使临床上可用的抗生素在对抗耐药菌方面存在一定的局限性。近年来,抗菌药物开发进展缓慢,且部分上市抗生素药物在较短时间就出现了耐药菌性。估计到2050年全球因耐药菌导致的死亡人数可达到1000万,造成的经济损失可达1005万亿美元,细菌感染已经成为紧随缺血性心脏病之后的人类第二大致死因素。为此,迫切需要寻找新型的治疗药物来应对耐药菌造成的感染性疾病。
2、抗菌肽(amp)是一类宿主防御肽,可通过多种途径杀伤细菌,如菌膜裂解、氧化损伤、抑制生物被膜形成等,不涉及特定蛋白的结合,不易产生耐药性,可对抗传统抗生素无效的细菌感染,因此amp具有作为治疗耐药菌感染性疾病的新一代抗生素的潜力。但大部分amp为线性多肽,在体内易被蛋白酶降解,结构不稳定,极大地限制其临床应用。前人通过对amp进行化学改造,如骨架改变、d型和非天然氨基酸替换等,提高了多肽的结构稳定性并取得了一定的进展。但在过去的几十年中,amp的结构稳定性仍是阻碍该类药物进入临床应用的重要因素,因此亟需寻找更有效地策略对amp进行改造,从而更好地解决耐药菌感染的问题。
3、ll-37是主要的人类amp之一,在防御局部和全身感染方面发挥重要作用。2018年,nibbering组基于人类ll-37改造的抗菌肽saap-148(slp-0),含有24个残基,具有α螺旋结构,相比模型肽ll-37,其在体内外表现出较高的抵抗耐药菌的能力。但作为直链多肽易被酶解,结构不稳定,抗耐药菌活性有限,对金黄色葡萄杆菌、铜绿假单胞菌及大肠杆菌的抑制均需要较高浓度才能实现,阻碍其药物开发。
技术实现思路
1、本发明的目的是针对现有技术中的不足,提供一种提高抗耐药菌活性及稳定性的订书肽。
2、本发明的另一的目的是,提供所述订书肽的制备方法。
3、本发明的另一的目的是,提供所述订书肽的用途。
4、为实现上述第一个目的,本发明采取的技术方案是:
5、一种订书肽,所述订书肽选自下列中的一种:
6、a)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基1l和5w被s5替换并环合;
7、b)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基4v和8v被s5替换并环合;
8、c)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基5w和9f被s5替换并环合;
9、d)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基7r和11l被s5替换并环合;
10、e)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基8v和12l被s5替换并环合;
11、f)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基9f和13k被s5替换并环合;
12、g)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基11l和15y被s5替换并环合;
13、h)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基12l和16w被s5替换并环合;
14、i)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基14r和18q被s5替换并环合;
15、j)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基15y和19l被s5替换并环合;
16、k)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基16w和20k被s5替换并环合;
17、l)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基18q和22p被s5替换并环合;
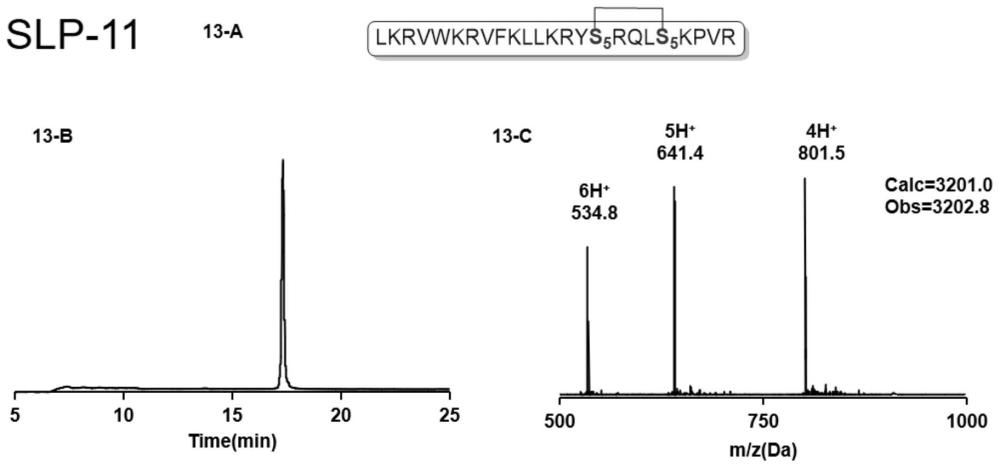
18、m)以ac-lkrvwkrvfkllkrywrqlkkpvr-nh2为肽链模板,其中氨基酸残基19l和23v被s5替换并环合。
19、本发明中模板多肽及改造的订书肽的序列如下表:
20、表1本发明中模板多肽及改造的订书肽的序列
21、
22、为实现上述第二个目的,本发明采取的技术方案是:
23、上述的订书肽的制备方法,包括如下步骤:
24、(1)以rink amide mbha氨基树脂为载体,其载样量为0.30mmol/g,在活化剂活化的dic/oxyme缩合剂的作用下分别使c端首个氨基酸与固相载体偶联;
25、(2)使用脱保护试剂脱去氨基酸上的fmoc保护基;
26、(3)使用缩合剂进行氨基酸的缩合;
27、(4)重复进行保护基的脱除-氨基酸缩合的操作,按照氨基酸序列合成肽链;其中,环合位点以s5来取代i和i+4位氨基酸;
28、(5)最后一个氨基酸使用脱fmoc保护基后,然后进行乙酰化;
29、(6)在环合剂的作用下使i和i+4位s5氨基酸发生烯烃复分解反应,合成订书肽;
30、(7)使用切割试剂将肽链从载体上切割下来,通过反相高效制备液相纯化得相应订书肽。
31、其中,作为本发明的另一优选例,步骤(7)中所述反相高效制备液相的色谱条件如下:色谱柱:ultimatexb-c18柱;流动相:流动相a为0.1%tfa/乙腈,流动相b为0.1%tfa/水;梯度洗脱程序:90%b洗脱0~3min,90%b~50%b洗脱40min;流速为8ml/min,进样体积为3ml,检测波长214nm和254nm。
32、作为本发明的另一优选例,步骤(1)中所述的缩合剂为dic-oxyme缩合体系,缩合体系的活化剂为dic,以dmf为溶剂。
33、作为本发明的另一优选例,步骤(2)中所述脱保护试剂为哌啶和dmf的混合溶液,所述哌啶和dmf的体积比为1:4。
34、作为本发明的另一优选例,骤(5)中所述乙酰化步骤的乙酰化试剂为ac2o,diea,dmf的混合液,所述ac2o,diea,dmf的体积比为1:1:8。
35、作为本发明的另一优选例,步骤(6)中所述环合剂为grubbsⅰ试剂的1,2-二氯乙烷的溶液,所述grubbsⅰ试剂与1,2-二氯乙烷的用量比为60:7,单位为mg/ml。
36、作为本发明的另一优选例,步骤(7)中,所述切割试剂为tfa、h2o、苯酚和tips的混合溶液,所述tfa、h2o、苯酚和tips的体积比为88.75:0.5:0.5:0.25;所述切割试剂与所述载体用量比为1:20,单位为ml/mg。
37、作为本发明的另一优选例,步骤(1)中固相合成时,固相树脂的载样量为0.30mmol/g。
38、作为本发明的另一优选例,步骤(1)中偶联反应的温度为50~60℃,优选反应温度为60℃;偶联反应的时间为20-30min,优选反应时间为30min。
39、作为本发明的另一优选例,步骤(2)中,脱fmoc保护是采用保护试剂反5min后,再次反应5min;脱除fmoc基团的反应温度为20~40℃,更优选为37℃。
40、作为本发明的另一优选例,s5后所接的第一个氨基酸反应时间为1h并按相同条件重复反应一次再进行下一步操作。
41、作为本发明的另一优选例,步骤(5)所述乙酰化是树脂在乙酰化试剂中反应15min,然后再次反应15min;反应温度为20~40℃,优选温度为37℃。
42、作为本发明的另一优选例,步骤(6)中所述环合剂为grubbsⅰ试剂的1,2-二氯乙烷的溶液,投料比为固相树脂载样量:grubbsⅰ:1,2-二氯乙烷=0.35:60:7(mmol/mg/ml)。
43、作为本发明的另一优选例,步骤(6)中所述环合是树脂在环合试剂中于恒温振荡器中反应两次,每次2h;反应温度为20~40℃,优选温度为37℃。
44、作为本发明的另一优选例,步骤(7)中,切割的温度为20~40℃,更优选为37℃;切割的时间为3h。
45、上述的订书肽在制备抑制金黄色葡萄球菌、大肠杆菌、铜绿假单胞菌及鲍曼不动杆菌药物中的应用。
46、本发明中,涉及的缩略词解释如下:
47、fmoc:芴甲氧羰基;dcm:二氯甲烷
48、dce:1,2-二氯乙烷
49、dmf:n,n-二甲基甲酰胺
50、oxyme:ethyl cyanoglyoxylate-2-oxime
51、dic:n,n-二异丙基碳二亚胺
52、s5:2-amino-2-methylhept-6-enoic acid
53、tfa:三氟乙酸
54、tips:三异丙基硅烷
55、grubbsⅰ:苯基亚甲基双(三环已基磷)二氯化钌
56、ms:质谱
57、hr-q-tof-ms:高分辨基质辅助激光解析电离飞行时间质谱。
58、本发明优点在于:
59、1、本发明提供了系列新的订书肽,通过对saap-148(slp-0)进行结构改造得到新的订书肽,使其具有更高的抗耐药菌活性,药理实验表明,本发明的订书肽可显著提高对金黄色葡萄球菌、大肠杆菌、铜绿假单胞菌及鲍曼不动杆菌的抑制活性,在临床耐药菌感染等相关疾病治疗中具有潜在的应用价值。
60、2、本发明以rink amide mbha氨基树脂作为固相载体,根据模板多肽slp-0:ac-lkrvwkrvfkllkrywrqlkkpvr-nh2氨基酸序列进行修饰改造,在dic-oxyme的缩合体系中,通过fmoc固相合成法,合成得到肽链,在保留关键氨基酸残基的基础上,在i,i+4氨基酸位置用s5代替原有氨基酸,直链肽在固相上合成完毕后,grubbsⅰ混合溶液加入到固相载体上,进行烯烃复分解反应环合,通过tfa混合溶液将目标订书肽切割下来,所得化合物通过反相高效液相进行纯化,采用hplc及ms等光谱进行表征分析。该方法简便易行,所得订书肽纯度大于95%,产率高。
61、3、为提高多肽的稳定性和抗耐药菌活性,本专利通过对slp-0进行订书化改造,在保留关键残基的基础上,以i,i+4方式进行设计,在肽链的非关键残基位置以s5分别替代i和i+4位氨基酸,环合后得到结构稳定的订书肽。共合成了13条订书肽(slp-1—slp-13),筛选得到的多肽提高了抗菌活性,能够对有害耐药细菌产生抑制性的正向调控。其中,slp-9对金黄色葡萄杆菌的抑菌效果得到显著提升,slp-12及slp-13对铜绿假单胞菌和大肠杆菌两个菌种的抑制效果有明显的增强。
- 还没有人留言评论。精彩留言会获得点赞!