一种手性合成(1R,2S)-贝达喹啉的方法与流程
本发明属于药物合成领域,具体涉及到使用手性诱导剂手性合成(1r,2s)-贝达喹啉的方法。主要使用廉价易得的手性诱导剂,定向催化提高非对映异构体的比例,降低原料药成本。
背景技术:
1、肺结核是由结核分支杆菌引起的肺部疾病,由于该疾病治疗时间长,还与自身免疫系统关系密切,很容易产生耐药性,故目前治疗方案分为一线用药(异烟肼、利福平、乙胺丁醇等),二线用药(喹诺酮类、环丝氨酸、阿米卡星等)和三线用药(贝达喹啉、德拉马尼等)。常规治疗是:先使用一线药物治疗6个月,如果未治愈,则需要二线药物和三线药物治疗长达两年或者更长的时间,而这些药物药效低,毒性大,价格也相对昂贵。根据世卫组织who对2022年结核病病例统计,目前结核病(tb)的数量尤其是多耐药性肺结核的病例在下降,但贝达喹啉仍然是临床确切需要的三线用药。
2、贝达喹啉(bedaquiline),化学名为1-(6-溴-2-甲氧基喹啉-3-基)-4-二甲氨基-1-苯基-2-(1-萘基)-2-丁醇。2012年12月美国fda加速批准了强生公司用于治疗耐药性肺结核的药物富马酸贝达喹啉,手性结构为1r,2s,其作用机理是通过抑制结核分枝杆菌的atp合成酶来杀死结核分枝杆菌。该药成为近20多年来首个具有全新作用机制的抗tb药物,同时也是有史以来首个明确用于多重耐药结核菌(mdr-tb)的抗tb的药物。其治愈率大于50%,但仍需要和其他作用机制的药物联合用药。
3、贝达喹啉有四个异构体,两个手性中心,但只有1r,2s构型具有有效活性。原研1r,2s-富马酸贝达喹啉的制备方法(cn 101547904 a)在发明专利较长的手性合成路线的基础上,公开了拆分法获得(1r,2s)-贝达喹啉的制备方法:6-溴-3-苄基-2-甲氧基喹啉在低温下,由二异丙基氨基锂(lda)脱去苄基的质子后,与3-二甲胺基-1-萘基-1-丙酮缩合成贝达喹啉,再经手性拆分得到单一化合物——(1r,2s)-贝达喹啉。但该反应中四种光学异构体的比例基本达到1:1:1:1,所以收率直接以25%计算,全路线收率不到10%。
4、另有报道,如shibasaki,catalytic asymmetis synthesis of r207910,j,amchem soc 2010 132:7905-7907报道了以不对称催化的方法构建第一个手性碳分子,再以不对称合成第二个手性碳分子的方法,但这条路线大于13步,整体收率也在10%左右,用到各类昂贵的催化剂,不适用于工业化生产。
5、国内也有相关制备专利的申请,如cn105175329a,以6-溴-3-苄基-2-甲氧基喹啉为起始物料,先与金属镁制成格式试剂,再与1-萘甲醛加成,也是多步骤的反应,但合成产物中四个手性异构体的具体比例未提及。国内专利cn106866525a报道了以n-苄基-脯氨醇锂及类似物为手性诱导剂,诱导(1r,2s)-贝达喹啉的生成比例,但使用的诱导剂市售较贵,而且还需要临用新制,后面仍需要拆分化合物,总体成本和直接制备贝达喹啉再经拆分相仿。
技术实现思路
1、为解决上述存在的技术问题,本发明提供一种手性合成(1r,2s)-贝达喹啉的方法,筛选出廉价的手性诱导剂,降低成本的同时可有效提高rs和sr这对异构体的比例。
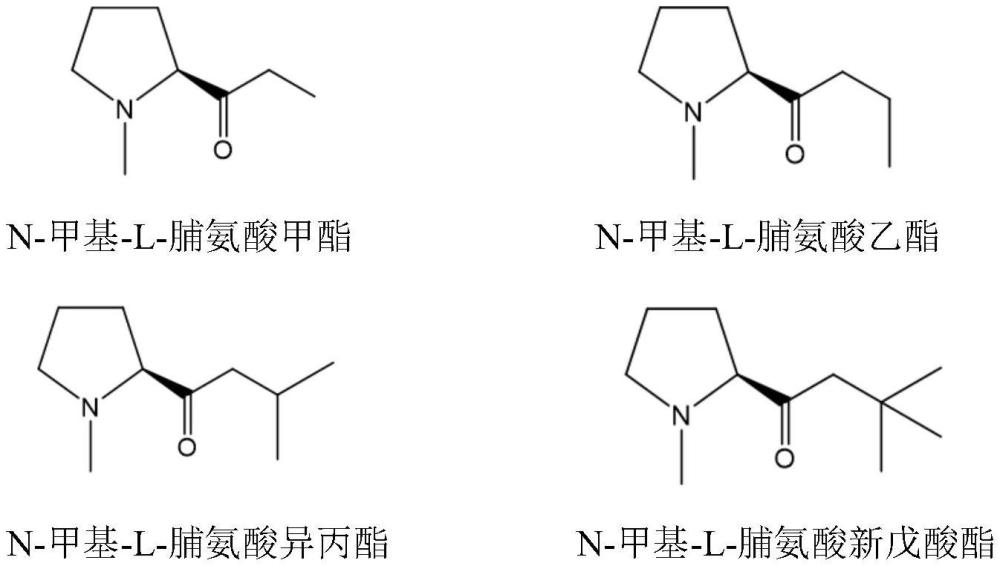
2、为实现上述目的,本发明采用如下技术方法:一种手性合成(1r,2s)-贝达喹啉的方法,在手性诱导剂的催化和诱导下,由二异丙基氨基锂作强碱,促进6-溴-3-苄基-2-甲氧基喹啉和3-二甲氨基-1-萘基-1-丙酮发生缩合反应,所得产物经拆分获得(1r,2s)-贝达喹啉;所述手性诱导剂选自n-甲基-l-脯氨酸甲酯、n-甲基-l-脯氨酸乙酯、n-甲基-l-脯氨酸异丙酯和n-甲基-l-脯氨酸新戊酸酯的一种或二种以上的组合。所述手性诱导剂的结构式如下:
3、
4、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,所述手性诱导剂选自n-甲基-l-脯氨酸乙酯。
5、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,包括如下步骤:
6、1)在氮气保护下,将二异丙基氨基锂和四氢呋喃加入到四口反应器中,降温到-80~-60℃,然后缓慢滴加手性诱导剂的四氢呋喃溶液,滴毕,继续于-80~-60℃下搅拌反应2小时;
7、2)向步骤1)所得反应体系中缓慢滴加6-溴-3-苄基-2-甲氧基喹啉的四氢呋喃溶液,滴毕,继续于-80~-60℃下搅拌反应2小时;
8、3)向步骤2)所得反应体系中缓慢滴加3-二甲胺基-1-萘基-1-丙酮的四氢呋喃溶液,滴毕,继续于-80~-60℃下搅拌反应14小时;
9、4)反应结束后向反应液中加入少量冰醋酸淬灭反应体系,缓慢升至室温,过滤,将滤液浓缩至粘稠状,加入乙醇分散体系2小时,过滤得粗品;
10、5)将粗品和r-联萘酚磷酸酯悬浮于丙酮中,于60~70℃搅拌2h后降温至室温,过滤,滤饼不经干燥直接加至2n碳酸钾溶液中,室温搅拌2h,过滤经纯化水洗涤,获得(1r,2s)-贝达喹啉。
11、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,按摩尔比,6-溴-3-苄基-2-甲氧基喹啉:手性诱导剂=1:0.1~2.0。
12、更进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,按摩尔比,6-溴-3-苄基-2-甲氧基喹啉:手性诱导剂=1:1.2。
13、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,步骤1)中,所述缓慢滴加是,控制滴加时间不少于1小时。
14、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,步骤2)中,所述缓慢滴加是,控制滴加时间不少于2小时。
15、进一步的,上述的一种手性合成(1r,2s)-贝达喹啉的方法,步骤3)中,所述缓慢滴加是,控制控制时间不少于3小时。
16、本发明,首先建立催化体系,在低温-80~-60℃下,将lda和手性诱导剂分别加入到四氢呋喃中,再缓慢加入6-溴-3-苄基-2-甲氧基喹啉的四氢呋喃溶液和3-二甲胺基-1-萘基-1-丙酮的四氢呋喃溶液,生成的产物为四个光学异构体的混合物。合成路线如下:
17、
18、反应结束后,首先使用冰醋酸淬灭反应体系,升至室温,加入乙醇分散得到产物对映异构体的粗品,最后经拆分得到单一构型的产物。
19、本发明的有益效果是:
20、本发明中,在反应前lda和手性诱导剂需要建立诱导体系,形成立体结构的配体,再在lda作用下脱去6-溴-3-苄基-2-甲氧基喹啉的苄基氢的同时,将该立体构型导入到6-溴-3-苄基-2-甲氧基喹啉的分子结构中,形成过渡态体系,最后经缩合后提高产品对映异构体对的产出比例。
21、本发明,采用高效液相(hplc)测定反应过程中四个手性光学异构体的比例。在没有手性诱导剂的存在下,两对非对映异构体的量比为近似1:1,其中每对非对映异构体中的对映异构体比例为1:1,与文献报道一致。本发明的目的就是提高产品对的生成比例,在使用本专利中的手性诱导剂后,经高效液相对反应体系的检测发现,产品一对非对映异构体比另外一对非对映异构体的比例大约为4:1,有效提高反应收率,且不经纯化水处理,直接经浓缩乙醇分散后,即可得到粗品,有效缩短生产时间。
22、本发明,筛选出四种同类型的手性诱导剂,可有效提高rs和sr这对异构体的比例,所采用的催化剂廉价易得,成本更低。
- 还没有人留言评论。精彩留言会获得点赞!