一种基于基因组学技术筛选轻木生长-防御候选基因的方法
本发明涉及生物,更具体的说是涉及一种基于基因组学技术筛选轻木生长-防御候选基因的方法。
背景技术:
1、轻木(学名:ochroma lagopus)又名百色木、巴沙木,是锦葵科轻木属的一种。属中等大小的常绿乔木,它的生长速度快,一年就可以长到五六米的高度,能够达到5-13厘米粗,3到4年即可采伐。不同于传统速生木材,轻木具有成熟期短、材质轻、孔隙率高和细胞腔大等显著优势,是风力发电机叶片制造的关键材料。研究人员发现,在外界胁迫条件下,由于自身资源限制,植物会进化出一套复杂精细的调控机制来介导生长和防御的平衡,以此实现生存效率的最大化。
2、近年来,研究发现,轻木在低温环境下会降低生长活性,且轻木极易受到害虫的侵蚀,一些生长-防御关键基因的收缩致使轻木具有较低的抗逆性能。
3、自然环境中各种生物和非生物胁迫是影响产量的巨大威胁。随着现代分子生物学的发展,从分子水平研究树种抵御逆境的机理已成为生态林业研究的一个重要任务,分子遗传学与生态学的整合诞生了生态基因组学即用基因组学的技术和手段研究生态林业领域的问题。通过全基因组数据的整理和分析,基于功能基因组学和结构基因组学进而比较不同物种间的基因组差异和相关性,可分析逆境响应相关基因的收缩。目前,尚未见到基于轻木全基因组学探究轻木的防御相关基因的研究报道。
4、因此,提供一种基于基因组学技术筛选轻木生长-防御候选基因的方法是本领域技术人员亟需解决的问题。
技术实现思路
1、有鉴于此,本发明提供了一种基于基因组学技术筛选轻木生长-防御候选基因的方法,通过全基因组测序和基因家族收缩分析筛选鉴定得到相关基因,为提高轻木抗逆能力实现本土化引种提供科学参考。
2、为了实现上述目的,本发明采用如下技术方案:
3、一种基于基因组学技术筛选轻木生长-防御候选基因的方法,具体步骤如下:
4、a.取轻木叶片进行液氮速冻研磨,提取高纯度高完整度的dna样品,进行文库构建和染色体级别基因组组装;
5、b.再利用orthomcl、paml、café软件进行基因家族聚类、分歧时间估计、基因家族收缩和分析,进一步筛选获取与轻木生长-防御相关的候选基因。
6、进一步,步骤a的具体操作步骤包括:
7、a-1.植物材料与叶片dna提取:于福建漳州大山林场取轻木叶片,液氮研磨、dna溶解、蛋白质去除、dna沉淀和洗涤处理,获取高质量的叶片dna样品,构建文库进行测序;
8、a-2.测序完成后,利用contigs数据使用hi-c平台进行染色体级别的辅助组装。
9、进一步,步骤a-1的具体操作步骤包括:
10、a.取轻木叶片在液氮中速冻研磨,根据天根(qiagen)试剂盒的标准流程提取dna;
11、b.使用琼脂糖凝胶电泳技术和qubit fluorometer检测提取的dna的浓度;
12、c.对于样本的纯度和完整度分别采用nanodrop (implen, ca, usa)和 agilent2100方法进行检测;
13、d.提取检验合格的基因组dna后,通过超声波破碎仪随机打断,并进行末端修复、加ploy a尾、加测序接头、纯化dna和pcr扩增步骤构建文库,文库构建完成后送安诺优达检测公司使用illumina hiseq 2500平台进行双端测序。
14、进一步,步骤a-2的具体操作步骤包括:
15、使用单拷贝直系同源库中植物特有的 1,614 条基因(embryophy_odb10数据集),对组装的结果进行busco分析,以评估基因组的完整性。根据评估指标,选出最佳的组装结果的contigs,用于后续的hi-c辅助组装。
16、使用hi-c数据辅助基因组组装,对原始下机数据进行过滤,获得高质量的reads,然后将高质量的reads比对到基因组,提取两端均比对到基因组唯一位置的paired-endreads用于后续的分析。利用参考基因组酶切片段信息,过滤得到有效相互作用validpaired-end reads(paired-end reads落在不同的酶切片段),最后分析酶切片段相互作用并将初步组装的contig/scaffold级别的基因组组装到染色体级别。
17、进一步,步骤a所获得的dna样品进行两种方法的纯度检测,并通过基因组学技术获得收缩基因家族;在此基础上,步骤b的具体操作步骤为:利用othomcl进行基因家族鉴定,phyml软件进行物种系统发育树构建,以brmc方法进行分歧时间估计,基因家族收缩和分析,将收缩和分析得到的基因集和各种功能数据库进行比对,然后明确基因功能在进化过程中的变化。
18、进一步,所述基因家族鉴定的具体步骤包括:
19、①对各个物种的基因集进行过滤;首先,一个基因存在多个可变剪接转录本时,仅保留编码区最长的转录本用于进一步分析;其次,为了保证蛋白编码的可靠性,将编码蛋白质小于50 个氨基酸的基因排除;
20、②通过blastp获得所有物种蛋白序列之间的相似性关系;
21、③使用orthomcl软件(li et al., 2003)对比对结果进行聚类。
22、进一步,所述系统发育树构建及分歧时间估计的具体步骤包括:
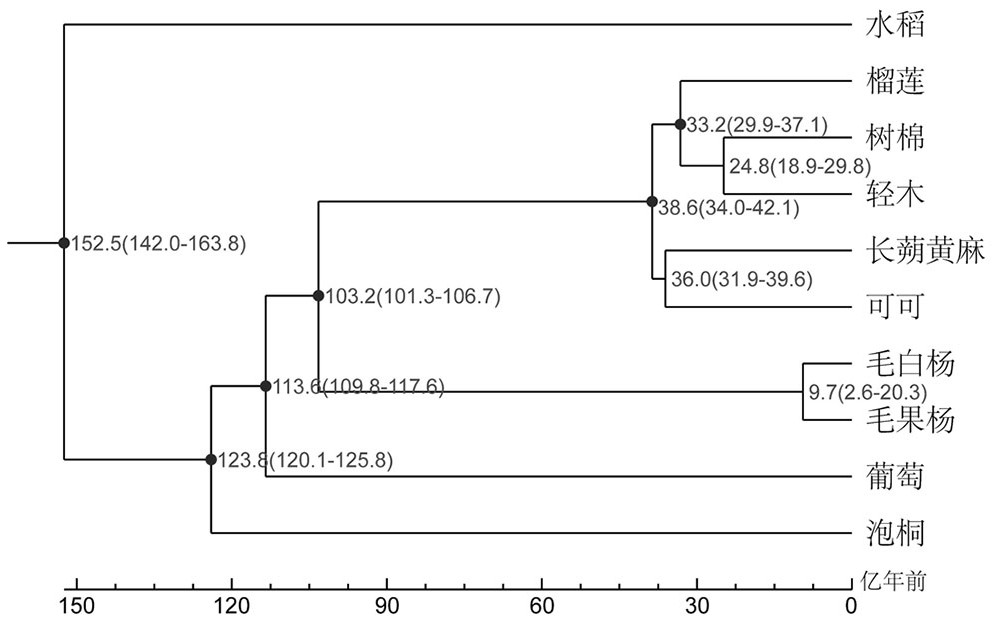
23、①通过软件muscle (http://www.drive5. com/muscle/) (edgar, 2004),对所有的单拷贝直系同源家族基因分别进行多序列比对,利用直系单拷贝同源基因的四重简并位点或选用直系单拷贝同源基因的全密码子序列,构建系统发育树,每个分支长度代表中性进化速率;所述单拷贝直系同源家族基因为在所有比对物种都有基因存在的基因家族,且各物种在此家族中的基因数为1;
24、②phyml软件(guindon et al., 2010)采用极大似然法进行物种系统发育树的构建(ml tree);
25、③先用标定时间加入到物种的系统发育树种,根据系统进化分析中整合的supergene序列,使用paml软件包中的mcmctree(http://abacus.gene.ucl.ac.uk/software/paml.html)软 件,以brmc方法进行分歧时间估计。
26、进一步,所述基因家族收缩和分析,将收缩和分析得到的基因集和各种功能数据库进行比对的具体步骤包括:
27、①根据基因家族的聚类分析结果,并过滤基因数在个别物种中存在异常的基因家族;
28、②使用café软件(http://sourceforge. net/projects/caféhahnlab/),利用pgm(probabilistic graphical models)模型对已知系统发育树 ,在指定的进化树下模拟基因的获得与丢失,并通过假设检验,进行基因家族和收缩分析,p参数设置为0.05;
29、③将鉴定到的收缩基因家族与nt、nr、blastx、blastp、pfam、eggnog相关数据库进行比对,获得序列的注释信息。
30、比较基因组分析是全基因组测序研究的重要组成部分,在完成组装注释完成之后,通过与近缘物种的蛋白序列比对,进行基因家族聚类等分析,从基因家族分析中挖掘物种的特有基因,探究特有基因与特殊生物学性状的关系。
31、物种基因家族收缩分析用于说明物种功能的减弱问题,根据基因家族的聚类分析结果,并过滤基因数在个别物种中存在异常的基因家族,基于具有分歧时间的进化树,使用café软件,利用pgm(probabilistic graphical models)模型对已知系统发育树,在指定的进化树下模拟基因的获得与丢失,并通过假设检验,估算经历收缩的基因家族的数量(定义显著或收缩的标准是 p 值<0.05和viterbi p<0.05),通过基因功能的注释得到生长-防御模块相关候选基因。
32、优选的,基因组学的研究可以正确处理海量数据,利用café软件进行收缩和分析可以确定生物间表型差异背后的遗传变化和导致变化的进化压力。
33、进一步,上述所述的方法在筛选轻木生长-防御候选基因中的应用。
34、进一步,步骤b之后可利用go富集、kegg富集、基因表达量分析等方法对所筛选出的生长-防御相关候选基因进行进一步功能分析。
35、进一步,步骤b之后可进一步利用rt-pcr技术对所筛选出的生长-防御相关候选基因进行验证。
36、为了明确轻木生长-防御相关基因的变化,本研究基于轻木的低抗逆性的表型,利用全基因组数据,通过基因家族收缩分析,鉴定与轻木生长-防御相关的候选基因,为轻木低防御的遗传解析和轻木的本土化繁育提供了理论依据和重要参考。
37、经由上述的技术方案可知,与现有技术相比,本发明公开提供了一种基于基因组学技术筛选轻木生长-防御候选基因的方法,能快速有效地筛选到与轻木生长-防御相关联的候选基因;本发明很好地利用了多种基因组研究手段来研究与轻木生长-防御相关的候选基因,弥补了轻木相关基因的研究空白;有利于轻木的本土化引种和遗传转化,具有良好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!