一种α-地贫分型试剂盒及其分型方法与流程
本发明涉及生物,尤其涉及一种α-地贫分型试剂盒及其分型方法。
背景技术:
1、α-地中海贫血(简称α-地贫)是由于α基因缺失或者突变,使得α肽链合成量下降,导致无效造血和红细胞破坏的一种溶血性贫血,是世界范围内高发的单基因遗传病,大约5%的世界人口携带α-珠蛋白基因的变异。α肽链基因位于人类第16号染色体上,在每条染色体上各有一对控制合成α链的α基因,因此每个细胞内有4个α基因,可发生不同程度(1~4个)基因异常;正常时为:αα/αα,静止型:-α/αα;轻型:-α/-α或--/αα;中间型:-α/--;重型:--/--。静止型和轻型无症状;中间型(hbh病)患儿出生时无明显症状,婴儿期以后逐渐出现贫血、疲乏无力、肝脾肿大、轻度黄疸,年龄较大患者可出现类似重型β-地中海。
2、贫血的特殊面容。合并呼吸道感染或服用氧化性药物、抗疟药物等可诱发急性溶血而加重贫血,甚至发生溶血危象;重型(hbbarts胎儿水肿综合征)胎儿常于30~40周时流产、死胎或娩出后半小时内死亡,胎儿呈重度贫血、黄疸、水肿、肝脾肿大、腹水、胸水。
3、α-地贫是一种常染色体隐性遗传,通常由hba1和hba2基因的缺失引起。hba1/hba2基因定位于16染色体(16p13.3),有3个外显子,由141个氨基酸组成,约29kb;中国人群常见的6种α地中海贫血基因突变位点,包括:非缺失α-地贫3种(c.369c>g、c.377t>c、c.427t>c),缺失α-地贫3种(--sea、-α4.2、-α3.7)。
4、α地贫目前该病尚无理想的治疗方法,研究表明在地贫高发区,对夫妇双方均为地贫基因携带者的孕妇在妊娠早期筛查或中期应用相应分析技术通过遗传筛查和产前基因诊断选择性地淘汰重症α-地贫胎儿则是控制该病发生的首要途径和有效预防措施。因此,对于缺失型α-地贫的检测急需一种简单、能快速准确基因分型的技术手段。
5、目前对α-地贫筛查方法如血常规参数检测分析、红细胞渗透脆性试验、血红蛋白电泳试验等特异性不高,较易产生假阴性。对于α-地中海贫血的分子诊断,southernblot杂交技术是金标准,准确性高,但操作繁琐、标本用量大、费时费力、检测通量小而不适合常规检测;其他taqman探针技术、变性高效液相色谱(dhplc)、dna直接测序和dna微阵列技术等。但这些方法检测过程繁琐、检测所用时间长、使用试剂或者仪器昂贵和特殊的标记引物等,因而不利于临床广泛的推广应用。目前市场上检测缺失型α-地贫多采用gap-pcr,该方法通过设计与缺失序列两侧序列互补的引物,缺失使本来在正常dna序列中相距很远的这对引物之间的距离因断端连接而靠近,并能扩增出特定长度的片段。再通过琼脂糖凝胶电泳,根据电泳片段大小检测样品的基因型,是缺失型地中海贫血检测的有效方法;然而该方法需要单独设计引物与单独扩增,操作繁琐,检测成本高,不利于临床广泛推广。
6、目前,缺少一种能够实现--sea、--thai、-α3.7、-α4.2长片段缺失检测与分型的简单快速的方法。
技术实现思路
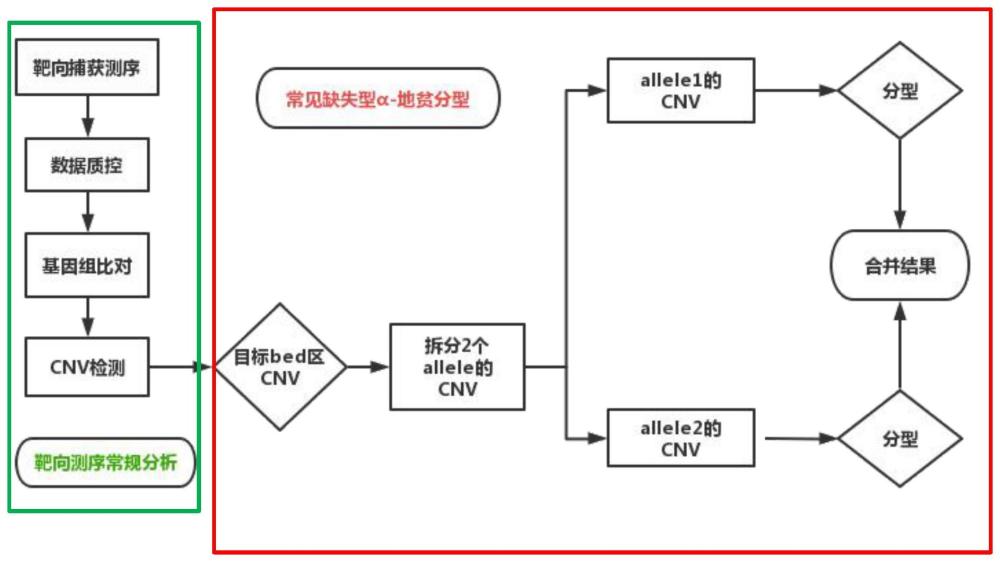
1、有鉴于此,本发明提供了一种缺失型α-地贫分型试剂盒及其分型方法。本发明提供的缺失型α-地贫分型试剂盒包括针对特定目标区域设计靶向捕获探针,利用该捕获探针构建文库测序获得测序数据,利用该测序数据和人类参考基因组获取目标区域的拷贝数变异结果,依次经allele拆分、分型即可实现对常见缺失型α-地贫的精准分型,该方法简单快速且分型结果准确可靠。
2、为了实现上述发明目的,本发明提供以下技术方案:
3、基因片段作为检测标志物在制备缺失型α-地贫分型试剂盒中的应用,所述基因片段选自如下基因:hs-40、nprl3、hbz、hbm、hbap1、hba2、hba1、hbq1和luc7l。
4、检测基因片段的试剂在制备缺失型α-地贫分型试剂盒中的应用,所述基因片段选自hs-40、nprl3、hbz、hbm、hbap1、hba2、hba1、hbq1和luc7l基因。
5、其中,所述缺失型α-地贫包括--sea缺失型、--thai缺失型、-α3.7缺失型和-α4.2缺失型。
6、一些实施方案中,所述试剂包括靶向捕获探针。
7、具体地,所述目标捕获探针覆盖的目标区域如表1所示的分型特异性补充区。
8、本发明还提供了检测基因片段的试剂,包括:以表1所示的至少一个分型特异性补充区为靶标设计得到的目标捕获探针。
9、本发明还提供一种缺失型α-地贫分型的方法,包括:
10、(1)结合人类参考基因组序列,获取表1所示的目标区域的拷贝数变异信息;
11、(2)将步骤(1)中与缺失型α-地贫常见类型相关的目标区域及其变异结果提取出来,对基因区域进行基因注释,对变异结果进行allele拆分,获得allele拆分结果;
12、(3)根据连续拷贝数缺失区域注释的基因以及缺失区域的长度对步骤(2)获得的allele拆分结果进行单个allele分型,合并2个allele的分型结果,获得缺失型α-地贫的型别。
13、一些实施方案中,步骤(1)具体包括:
14、①将样本dna打断,利用权利要求5所述的靶向捕获探针进行捕获,利用捕获的目标片段构建测序文库,对测序文库进行测序;
15、②将测序数据中的测序接头以及低质量碱基和/或序列去除,评估原始数据的质量;
16、③使用bwa软件将步骤②处理后的测序数据比对到人类参考基因组上,获得存储有比对信息的bam文件;
17、④对所述bam文件进行排序、局部重排以及bqsr校正处理,获得处理后的bam文件;
18、⑤将所述处理后的bam文件进行拷贝数变异分析检测,获得目标区域的拷贝数变异结果。
19、一些实施方案中,步骤(2)中,所述缺失型α-地贫的常见类型包括--sea缺失型、--thai缺失型、-α3.7缺失型和-α4.2缺失型。
20、一些实施方案中,步骤(2)具体包括:
21、将步骤(1)中与缺失型α-地贫常见类型相关的目标区域及其变异结果提取出来,对目标区域进行基因注释,对变异结果进行allele拆分,获得allele拆分结果;
22、所述allele拆分针对2拷贝缺失的区域、2拷贝缺失+1拷贝缺失的组合区域进行拆分;其中,2拷贝缺失记为cn0,1拷贝缺失标记为cn1;
23、所述异常处理的方法包括:
24、a.当单个bed区域存在拷贝数缺失,但其前边相邻的两个区域和后边相邻的两个区域的拷贝数均正常,则判定该区域拷贝数正常;
25、b.当单个bed区域存在拷贝数缺失,但不符合a,则跳过该区域,直至出现连续的拷贝数缺失区域;
26、所述allele拆分的方法包括:
27、c.将连续的cn0区域,拆分为cn1+cn1;
28、d.连续的cn0+cn1区域,根据cn0的长短进行拆分:
29、(1)如果cn0长度大于等于5个,则将cn0区域看作单独allele上的cn1缺失;
30、(2)如果cn0长度小于5个,则将cn0区域看作两个不同allele上同时缺失的区域。
31、步骤(3)中,所述单个allele分型的方法包括:
32、i.如果hbz_1和hbq1_3基因缺失,并且缺失的bed区域大于等于35个,则为--thai型2拷贝缺失;
33、j.如果hbm_1和hbq1_3基因缺失,hbz_3基因未缺失,且缺失的bed区域在25-32之间,则为--sea型2拷贝缺失;
34、k.如果hba2_3和hba2_hba1基因缺失,hbap1_hba2和hbq1_3基因未缺失,且缺失的bed区域在5-14之间,则为-α3.7型1拷贝缺失;
35、l.如果hbap1_hba2和hba2_3基因缺失,hba1_1和hbm_hbap1基因未缺失,且缺失的bed区域在5-13之间,则为-α4.2型1拷贝缺失;
36、m.如果hba1_1,hba1_2和hba1_3基因缺失,hba2_2和hba2_3基因缺失,则为--型2拷贝缺失;
37、n.如果hba1_1,hba1_2和hba1_3基因缺失,hba2_2和hba2_3基因未缺失,则为-α型1拷贝缺失;
38、o.如果hba1_1,hba1_2和hba1_3基因未缺失,hba2_2和hba2_3基因缺失,则为-α型1拷贝缺失;
39、p.如果a~g均不满足,则为αα型0拷贝缺失。
40、本发明针对对hba1/hba2及其上下游基因的区域设计得到靶向捕获探,利用该捕获探针构建文库测序获得测序数据,利用该测序数据和人类参考基因组获取目标区域的拷贝数变异结果,依次经allele拆分、分型,实现了--sea、--thai、-α3.7、-α4.2缺失型α-地贫的精准分型,同时也可将常规wes中加入该区域中非外显子探针整合成加强版wes,为新生儿筛查、遗传病分子诊断提供一种高性价比的检测技术。
- 还没有人留言评论。精彩留言会获得点赞!