杂环酰胺类化合物及其制备方法、药物组合物和应用
本发明涉及一种杂环酰胺类化合物及其制备方法、药物组合物和应用,尤其涉及一种具有akr1c3选择性抑制活性的杂环酰胺类化合物及其制备方法、药物组合物和应用。
背景技术:
1、人醛酮还原酶(human aldo-keto reductase family,akr)是氧化还原酶家族成员之一,目前已发现的家族有16个,包含151个成员,是一类主要存在于胞浆中的单体可溶性蛋白,分子量约为34~37kda。大多数akr成员的生理功能多为在体内催化氧化还原反应,利用nad(p)h作为辅酶,将内源性(类固醇、糖醛、活性脂醛、酮前列腺素、神经甾体、胆汁酸和视网膜)或外源性(药物,致癌小分子)底物结构中的醛或酮向一级或二级醇转化,也可催化硝基等基团的还原,属i相代谢酶。因为其在体内广泛的分布和底物来源,醛酮还原酶家族在甾体激素代谢合成、药物代谢与失活、氧化应激反应的调控、致癌物解毒等生命活动中发挥重要作用。在人体中,已发现的akr1c酶(aldo-keto reductase family 1member c)包括4个异构体,即akr1c1-akr1c4,这些异构体有着86%氨基酸序列同源性,但是具有不同的组织分布偏好和底物选择性,其在人体内介导不尽相同却又相互关联的生理功能,和多种疾病的发生发展有着千丝万缕的联系。其中akr1c3是当前生物表型,机理研究最透彻、“可药靶性”最高的成员之一。
2、akr1c3,也称为人17β-羟基类固醇5型脱氢酶(human 17β-hydroxysteroiddehydrogenase type 5,17β-hsd5)在体内与激素的代谢密切相关。akr1c3首先以作为前列腺素f合酶(prostaglandin f synthase,pgf),将前列腺素d2(prostaglandin d2,pgd2)转变为9α,11β-pgf2α和将前列腺素h2(prostaglandin h2,pgh2)转变为pgf2α,通过发挥酮类固醇还原作用,akr1c3能催化弱雄激素前体δ4-雄甾烯-3,17-二酮(4-androstene-3,17-dione,δ4-ad)和5α-雄甾烷-3,17-二酮(5α-androstane-3,17-dione)分别转化为强效雄激素睾酮和5α-二氢睾酮(5α-dihydrotestosterone,5α-dht),将弱雌激素雌酮还原为强雌激素17β-雌二醇,将强孕激素孕酮转化为弱孕激素20α-羟基孕酮。这些复杂的激素代谢功能预示着其与激素代谢有关的疾病有紧密的关联,尤其是激素依赖性的肿瘤,如前列腺癌,去势抵抗性前列腺癌,乳腺癌等。
3、另外,多项研究表明akr1c3可以影响人体内多条关键的信号通路,如丝裂原活化蛋白激酶(mitogen-activated protein kinase,mapk)、磷脂酰肌醇3激酶(phosphoinositide 3-kinase,pi3k)/蛋白激酶b(protein kinase b,akt)、细胞外调节蛋白激酶(extracellular signal-regulated kinases,erk)、核因子kappa-b(nuclearfactor kappa-b,nf-κb)、nf-e2相关因子2(nf-e2-related factor-2,nrf2)等,调控人体正常的生物信号网络,因此akr1c3还和许多非激素依赖性的疾病有关。
4、多项研究证实akr1c3的高表达与多种癌症的发生发展和预后不良有关,被认为是多种肿瘤潜在的诊断或预后的生物标志物。在各种治疗手段高度发达的今天,化疗依然是当前治疗肿瘤的主要手段之一。但是化疗容易耐药,给药量大,副作用大的缺陷依旧无法避免。akr1c3对于多种含羰基的外源性化合物,包括许多化疗药物的还原,以及对多条信号通路的影响可以导致抗肿瘤药物的解毒和耐药性的产生。尽管已经开发了许多akr1c3抑制剂,但尚未有化合物在临床实践中成功应用。因此,开发新型、强效且特异性akr1c3抑制剂具有潜在的应用前景。
技术实现思路
1、发明目的:本发明的第一目的是提供一种杂环酰胺类化合物,第二目的是提供一种所述化合物的制备方法,第三目的是提供一种包含所述化合物的药物组合物,第四目的是提供一种所述化合物或其药物组合物的应用。
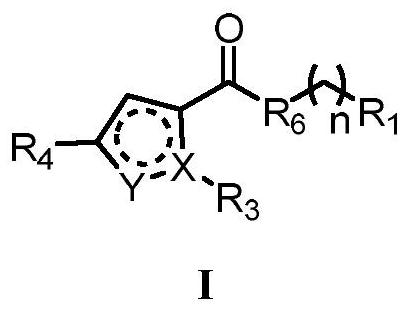
2、技术方案:本发明所述的杂环酰胺类化合物具有式i的结构,还包含其药学上可接受的盐:
3、
4、其中:
5、r1选自*-w-r2
6、w选自6-10元芳基、含有一个n、o、s的5-10元芳杂基、4-10元脂环基、苯并4-6元脂环基、苯并5-6元含有1-2个o的脂杂环基;
7、r2选自氢、c1-c4烷基、c1-c4烷氧基、羧基、硝基、氰基、卤素、c1-c4卤代烷基、c1-c4卤代烷氧基;
8、r3选自氢、c1-c4烷基;
9、r4选自h、
10、r5选自苯基、c1-c4烷基、3-6元环烷基,其中苯基被氢、c1-c4烷基、硝基、c1-c4烷氧基、卤素、氰基取代;
11、x选自n、o、s;
12、y选自c、o;
13、r6选自nh、nr7、哌嗪基;
14、r7选自c1-c4烷基;
15、n选自0、1、2。
16、优选,所述结构中:
17、r1选自*-w-r2:
18、w选自苯基、萘基、含有一个n的6元或9-10元芳杂基、含有一个n、o、s的5元芳杂基、6-10元脂环基、苯并5-6元含有2个o的脂杂环基;
19、r2选自氢、c1-c4烷基、c1-c4烷氧基、羧基、硝基、氰基、卤素、c1-c4卤代烷氧基;
20、r4选自h、
21、r5选自苯基、c1-c4烷基、3-5元环烷基,其中苯基被氢、c1-c4烷基、硝基、c1-c4烷氧基、卤素、氰基取代。
22、优选,所述结构中:
23、r1选自*-w-r2;
24、w选自苯基、吡啶基、呋喃基、噻吩基、吡咯基、环己基、金刚烷基、苯并二氧杂环戊烯基、萘基、吲哚基、喹啉基、异喹啉基;
25、r2选自氢、c1-c4烷烃、c1-c4烷氧基、羧基、硝基、氰基、卤素、c1-c4卤代烷基;
26、r3选自h、甲基、乙基;
27、r4选自
28、r5选自甲基、乙基、丙基、异丙基、环丙基、环丁基、环戊基、苯基,或者甲基、硝基、甲氧基、氟、氯、氰基取代的苯基;
29、x选自n、o、s;
30、y选自c、o;
31、r6选自nh、nch3、哌嗪基;
32、n选自1、2。
33、优选,所述结构中:
34、r2选自氢、甲基、甲氧基、氰基、羧基、硝基、三氟甲基、氟、氯、叔丁基、2,4,6位-三甲基取代基、3,4位-二甲氧基取代基、3,4,5位-三甲氧基取代基、2,4位-二氟取代基、2,4,6位-三氟取代基;
35、x选自n时,y选自c、o;
36、或者x选自o时,y选自c;
37、或者x选自s时,y选自c。
38、优选,选自以下任一的化合物:
39、
40、
41、其中,本发明所述的药学上可接受的盐为所述化合物与选自以下任一的酸形成的盐:
42、盐酸、氢溴酸、硫酸、磷酸、碳酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、柠檬酸、酒石酸、苹果酸、乳酸、丙酮酸、乙酸、马来酸、琥珀酸、富马酸、水杨酸、苯基乙酸、杏仁酸、阿魏酸。
43、“药学上可接受的盐”是指化合物的盐,由具有特定取代基的化合物与相对无毒的酸或碱制备。当化合物中含有相对酸性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的游离体形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机氨或镁盐或类似的盐。当化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的游离体形式接触的方式获得酸加成盐。药学上可接受的酸加成盐的实例包括无机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸(形成碳酸盐或碳酸氢盐)、磷酸(形成磷酸盐、磷酸一氢盐、磷酸二氢盐、硫酸(形成硫酸盐或硫酸氢盐)、氢碘酸、亚磷酸等;以及有机酸盐,所述有机酸包括如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸和甲磺酸等类似的酸;有机酸盐还包括氨基酸(如精氨酸等)、葡糖醛酸等有机酸的盐。当某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。优选地,以常规方式使盐与碱或酸接触,再分离母体化合物,由此再生化合物的游离体形式。化合物的游离体形式与其各种盐的形式的不同之处在于某些物理性质,例如在极性溶剂中的溶解度不同。
44、“药学上可接受的盐”可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。一般地,优选醚、乙酸乙酯、乙醇、异丙醇或乙腈等非水介质。
45、本发明所述的杂环酰胺类化合物的制备方法选自以下任一:
46、方法一:
47、
48、化合物1经酰化、水解,然后与化合物5缩合得到所述式i的化合物;
49、具体地,以2-吡咯甲酸甲酯衍生物1为起始原料,二氯甲烷作为溶剂,氯化铝为碱,与不同类型的酰氯2,发生傅克酰基化反应后,得到酰基吡咯酯中间体3,以甲醇为溶剂,少许四氢呋喃助溶,滴加氢氧化锂发生酯水解后,加酸调ph至1~2,得到羧酸中间体4,在dmf作溶剂,hatu为缩合剂,dipea为碱,和亚甲基个数为1、2取代的仲胺或经甲基取代的叔胺5,发生酰胺缩合后,得到式i的化合物。
50、方法二:
51、
52、以化合物7与对氰基苄胺经酰胺缩合后得到所述式i的化合物;
53、具体地,以五元杂环羧酸7为起始原料,与对氰基苄胺经酰胺缩合反应后,得到式i的化合物。
54、其中,r1、r3、r5、r6、x、y、n的定义如前所述。
55、将相应的酸与以上方法制备的化合物i成盐,即得所述杂环酰胺类化合物的药学上可接受的盐。
56、本发明所述的药物组合物包含本发明所述的杂环酰胺类化合物以及药学上可接受的载体。
57、所述药物的剂型选自胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂、贴剂。可任意混合的载体根据剂型、给药形式等可以改变。其中,药学上可接受的载体例如:赋形剂、粘合剂、崩解剂、润滑剂、矫味剂、香味剂、着色剂或甜味剂等。
58、“药学上可接受的载体”可为药物生产领域中广泛采用的辅料。辅料主要用于提供一个安全、稳定和功能性的药物组合物,还可以提供方法,使受试者接受给药后活性成分以所期望的速率溶出,或促进受试者接受组合物给药后活性成分得到有效吸收。所述的药用辅料可以是惰性填充剂,或者提供某种功能,例如稳定该组合物的整体ph值或防止组合物活性成分的降解。所述的药用辅料可以包括下列辅料中的一种或多种:粘合剂、助悬剂、乳化剂、稀释剂、填充剂、成粒剂、胶粘剂、崩解剂、润滑剂、抗粘着剂、助流剂、润湿剂、胶凝剂、吸收延迟剂、溶解抑制剂、增强剂、吸附剂、缓冲剂、螯合剂、防腐剂、着色剂、矫味剂和甜味剂。
59、所述药物可根据公开的内容使用本领域技术人员已知的任何方法来制备。例如,常规混合、溶解、造粒、乳化、磨细、包封、包埋或冻干工艺。
60、所述药物可以以任何形式给药,包括注射(静脉内)、粘膜、口服(固体和液体制剂)、吸入、眼部、直肠、局部或胃肠外(输注、注射、植入、皮下、静脉内、动脉内、肌内)给药。所述的药物还可以是控释或缓释剂型(例如脂质体或微球)。固体口服制剂的实例包括但不限于粉末、胶囊、囊片、软胶囊剂和片剂。口服或粘膜给药的液体制剂实例包括但不限于悬浮液、乳液、酏剂和溶液。局部用制剂的实例包括但不限于乳剂、凝胶剂、软膏剂、乳膏剂、贴剂、糊剂、泡沫剂、洗剂、滴剂或血清制剂。胃肠外给药的制剂实例包括但不限于注射用溶液、可以溶解或悬浮在药学上可接受载体中的干粉制剂、注射用悬浮液和注射用乳剂。所述的药物组合物的其它合适制剂的实例包括但不限于滴眼液和其他眼科制剂;气雾剂,如鼻腔喷雾剂或吸入剂;适于胃肠外给药的液体剂型;栓剂以及锭剂。
61、本发明所述的杂环酰胺类化合物或其药物组合物应用在制备akr1c3抑制剂药物中。
62、优选,所述药物为预防或治疗癌症、逆转肿瘤耐药的药物,具体用于治疗癌症的过度增殖、细胞生长抑制或细胞毒性。
63、有益效果:与现有技术相比,本发明具有如下显著优点:
64、本发明所设计的大部分化合物对akr1c3具有良好的抑制活性和选择性(达到微摩尔浓度水平、甚至是纳摩尔浓度水平),能够有效抑制肿瘤细胞增殖和有效逆转肿瘤耐药。丰富了现有akr1c3抑制剂的结构类型,应用于与akr1c3过表达相关疾病的治疗药物,特别是在抗肿瘤领域具有较好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!