吲哚啉酰基衍生物及异喹啉甲酰基衍生物的合成方法与流程
本发明属于精细有机化学合成领域,更具体的涉及吲哚啉酰基衍生物及异喹啉甲酰基衍生物的合成方法。
背景技术:
1、吲哚啉和异喹啉类衍生物作为手性源广泛用于医药、农药、新材料等科研领域。具有手性亚氨基酸结构特征的吲哚啉和异喹啉类化合物,作为起始原料,可以用来合成一系列衍生产物。吲哚啉衍生产物(s)-2-甲酰基-吲哚啉-1-羧酸叔丁酯和异喹啉衍生产物(s)-3-甲酰基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯,均具有良好的光学异构活性而日益受到业内重视。
2、目前公开的合成方法,均采取如下路线,即:方法a(s)-2-甲酰基-吲哚啉-1-羧酸叔丁酯合成路线;方法b(s)-3-甲酰基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯合成路线。
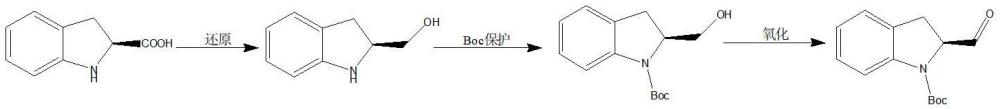
3、上述两种物质的合成,采用的路线极为相似,是目前业内合成此两类物质采取的普遍之法。即先通过对吲哚啉或者异喹啉的底物酸(羧基物)进行还原,获得吲哚啉或者异喹啉的羟甲基衍生物,再对吲哚啉或者异喹啉的亚氨基氮进行boc(叔丁氧羰基)保护,最后经氧化,得到吲哚啉或者异喹啉的甲酰基衍生物。
4、对于合成的第二步,由于吲哚啉和异喹啉环存在较大的位阻作用,因此,对这两类化合物的亚氨基氮进行 boc 保护,反应存在一定难度,需要消耗 较 长 的 时 间,收率也偏低(例如参见:lafzi,ferruh 等,design and synthesis of novel indoline-(thio)urea hybrids,synthetic communications,2019,vol.49,#24,p.3510-3527 以及us2014148452a1)。
5、对于合成的第三步,存在使用的氧化剂价格昂贵、反应后处理复杂等问题(例如参见:molander,gary a等,investigations concerning the organolanthanide and group3 metallocene-catalyzed cyclization –functionalization of nitrogen-containingdienes,tetrahedron,2005,vol.61,#10,p.2631-2643 和wo2009151626a1)。李先栓详细讨论了铜催化氧化醇生成醛的反应机理,但反应所需温度较高(李先栓,铜催化氧化乙醇生成醛的反应机理.化学教学,2011(9):45-46)。
6、因此,目前普遍采用的吲哚啉和异喹啉甲酰基衍生物的合成方法,存在反应难度大、原料成本高、后处理繁琐等问题,并不利于规模化生产。
技术实现思路
1、1、发明目的。
2、本发明提出了一种吲哚啉酰基衍生物及异喹啉甲酰基衍生物的合成方法。
3、2、本发明所采用的技术方案。
4、一种吲哚啉酰基衍生物的合成方法,其步骤包括:
5、(1)(s)-吲哚啉-2-羧酸经硼氢化钠还原,再加入甲醇反应,生成(s)-吲哚啉-2-甲醇;
6、(2)(s)-吲哚啉-2-甲醇与二碳酸二叔丁酯在超声辅助下进行反应,制得(s)-2-羟甲基-2,3-二氢吲哚-1-羧酸叔丁酯;
7、(3)用铜粉与氧化铜的混合物对(s)-2-羟甲基-2,3-二氢吲哚-1-羧酸叔丁酯进行氧化,氧化过程中用微波辅助反应,制得(s)-2-甲酰基-吲哚啉-1-羧酸叔丁酯。
8、优选的,步骤(1)具体为氮气保护下,将(s)-吲哚啉-2-羧酸溶解于四氢呋喃中,搅拌下分批加入硼氢化钠,加毕,滴入碘的四氢呋喃溶液,滴加毕搅拌至体系红色消失,继续升温回流;回流毕,冷至室温,滴加甲醇至无气泡产生;减压脱溶,加入二氯甲烷溶解残余物,以氢氧化钾水溶液洗涤有机相,分去水相,有机相以无水硫酸钠干燥后减压脱溶,所得羧基还原物粗品以甲苯重结晶,得到(s)-吲哚啉-2-甲醇。
9、优选的,所述步骤(1)中(s)-吲哚啉-2-羧酸与硼氢化钠的摩尔比例为1:2.4~1:2.5。
10、优选的,步骤(2)具体为将步骤(1)所得(s)-吲哚啉-2-甲醇溶解于甲醇中,分批加入二碳酸二叔丁酯,于超声反应器内,超声辅助反应进行,控制反应温度不超过28℃,反应至tlc监测原料消失;减压脱溶,得亚氨基氮boc保护产物(s)-2-羟甲基-2,3-二氢吲哚-1-羧酸叔丁酯。
11、优选的,步骤(2)中(s)-吲哚啉-2-甲醇与二碳酸二叔丁酯摩尔比为1:1~1:1.1,超声功率为200~400w,反应温度为20~28℃。
12、优选的,步骤(3)具体为将步骤(2)所得(s)-2-羟甲基-2,3-二氢吲哚-1-羧酸叔丁酯溶解于dmf中,加入铜粉与氧化铜的混合物,鼓入空气,微波辅助反应进行,控制反应温度在148~153℃之间;反应冷却至室温,滤去铜粉与亚铜粉固体残渣,将反应液滴入水中,以甲基叔丁基醚萃取,分去水层,所得有机相以无水硫酸钠干燥后减压脱溶,即得目标产物(s)-2-甲酰基-吲哚啉-1-羧酸叔丁酯。
13、优选的,所述步骤(3)中铜粉与氧化铜混合物与(s)-2-羟甲基-2,3-二氢吲哚-1-羧酸叔丁酯重量比为10%~30%,其中铜粉与氧化铜重量比为1:2~1:2.5,微波功率为400~500w,反应时间在1~2小时。
14、本发明还公开了一种异喹啉甲酰基衍生物的合成方法,其步骤包括:
15、(1)(s)-(-)-1,2,3,4-四氢异喹啉-3-羧酸经硼氢化钠还原,再加入甲醇反应,生成(s)-(-)-1,2,3,4-四氢异喹啉-3-甲醇;
16、(2)(s)-(-)-1,2,3,4-四氢异喹啉-3-甲醇与二碳酸二叔丁酯在超声辅助下进行反应,制得(s)-3-羟甲基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯;
17、(3)用铜粉与氧化铜的混合物对(s)-3-羟甲基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯进行氧化,氧化过程中用微波辅助反应,制得(s)-3-甲酰基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯。
18、优选的,步骤(1)具体为氮气保护下,将(s)-(-)-1,2,3,4-四氢异喹啉-3-羧酸溶解于四氢呋喃中,搅拌下分批加入硼氢化钠,加毕,滴入碘的四氢呋喃溶液,滴加毕搅拌至体系红色消失,继续升温回流;回流毕,冷至室温,滴加甲醇至无气泡产生;减压脱溶,加入二氯甲烷溶解残余物,以氢氧化钾水溶液洗涤有机相,分去水相,有机相以无水硫酸钠干燥后减压脱溶,所得羧基还原物粗品以甲苯重结晶,得到(s)-(-)-1,2,3,4-四氢异喹啉-3-甲醇。
19、优选的,所述步骤(1)中(s)-(-)-1,2,3,4-四氢异喹啉-3-羧酸与硼氢化钠的摩尔比例为1:2.4~1:2.5。
20、优选的,步骤(2)具体为将步骤(1)所得(s)-(-)-1,2,3,4-四氢异喹啉-3-甲醇溶解于甲醇中,分批加入二碳酸二叔丁酯,于超声反应器内,超声辅助反应进行,控制反应温度不超过28℃,反应至tlc监测原料消失;减压脱溶,得亚氨基氮boc保护产物(s)-3-羟甲基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯。
21、优选的,步骤(2)中(s)-(-)-1,2,3,4-四氢异喹啉-3-甲醇与二碳酸二叔丁酯摩尔比为1:1~1:1.1,超声功率为200~400w,反应温度为20~28℃。
22、优选的,步骤(3)具体为将步骤(2)所得(s)-3-羟甲基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯溶解于dmf中,加入铜粉与氧化铜的混合物,鼓入空气,微波辅助反应进行,控制反应温度在148~153℃之间;反应冷却至室温,滤去铜粉与亚铜粉固体残渣,将反应液滴入水中,以甲基叔丁基醚萃取,分去水层,所得有机相以无水硫酸钠干燥后减压脱溶,即得目标产物(s)-3-甲酰基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯。
23、优选的,所述步骤(3)中铜粉与氧化铜混合物与(s)-3-羟甲基-3,4-二氢-1h-异喹啉-2-羧酸叔丁酯重量比为10%~30%,其中铜粉与氧化铜重量比为1:2~1:2.5,微波功率为400~500w,反应时间在1~2小时。
24、3、本发明所产生的技术效果。
25、(1)本发明在合成的第二步,即合成亚氨基氮boc保护产物过程中,以超声辅助反应进行,可以有效的克服位阻影响,使得反应更为容易,缩短了反应时间,提高了反应收率。
26、(2)本发明在合成的第三步,即对化合物的羟甲基进行氧化阶段,以微波辅助铜粉/氧化铜催化反应的进行,可大大降低同类反应中醇羟基氧化成醛所需的温度,也避免了使用价格昂贵,后处理复杂的氧化剂。
27、(3)本发明操作简便、单步收率高、经济上可行,在同类化合物的路线选择与设计上具有明显的优势。
- 还没有人留言评论。精彩留言会获得点赞!