一种具有电光响应特性的光子晶体胶囊及其制备方法与流程
本发明属于功能材料技术领域,具体涉及一种具有电光响应特性的光子晶体胶囊及其制备方法。
背景技术:
光子晶体具备优异的光学性能,在显示、防伪、信息记录等领域都具有重要的作用。然而,由于这类物质容易受环境中水蒸气、二氧化碳、紫外线或化学蒸气等影响,物理和化学性质不稳定,使用寿命不长。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种光子晶体胶囊及其制备方法,本发明提供的光子晶体胶囊墨水性能稳定,使用寿命长。
本发明提供了一种具有电光响应特性的光子晶体胶囊的制备方法,包括以下步骤:
采用微流控技术,以由一维光子晶体和油溶性聚合前体组成的油相为分散相,以由表面活性剂和超纯水组成的水相为连续相,以由水溶性聚合前体、表面活性剂和超纯水组成的水相为收集相,油相中的油溶性聚合前体和收集相中的水溶性聚合前体在油水交界处发生界面聚合生成包裹光子晶体的聚合物胶囊壁,得到光子晶体胶囊墨水。
优选的,所述一维光子晶体的反射波长位于400~800nm。
优选的,所述一维光子晶体按照如下方法进行制备:
将向列相液晶与手性掺杂剂混合,得到一维光子晶体。
优选的,所述手性向列相液晶选自型号为bhr-59001、5cb或e7的手性向列相液晶,所述手性掺杂剂选自型号为s-811、r811、cb15或r5011的手性向列相液晶,所述手性掺杂剂占所述一维光子晶体的质量百分比为20.2wt%~28.6wt%。
优选的,所述油溶性聚合前体为四乙烯五胺,所述分散相中油溶性聚合前体的质量百分比为1%~5%。
优选的,所述连续相中的表面活性剂选自十二烷基硫酸钠或十二烷基苯磺酸钠,所述连续相中的表面活性剂的质量百分比为1%~5%。
优选的,所述收集相中的表面活性剂选自十二烷基硫酸钠或十二烷基苯磺酸钠,所述收集相中表面活性剂的质量百分比为1%~5%;所述收集相中的水溶性聚合前体为甲苯二异氰酸酯,所述收集相中的水溶性聚合前体的质量百分比为1%~5%。
优选的,所述分散相的流速1~4微升每分钟,所述连续相的流速为40~400微升每分钟,所述收集相在收集瓶中的体积为2~40毫升。
本发明还提供了一种上述制备方法制备得到的具有电光响应特性的光子晶体胶囊在显示、防伪或信息记录中的应用。
与现有技术相比,本发明提供了一种具有电光响应特性的光子晶体胶囊的制备方法,包括以下步骤:采用微流控技术,以由一维光子晶体和油溶性聚合前体组成的油相为分散相,以由表面活性剂和超纯水组成的水相为连续相,以由水溶性聚合前体、表面活性剂和超纯水组成的水相为收集相,油相中的油溶性聚合前体和收集相中的水溶性聚合前体在油水交界处发生界面聚合生成包裹光子晶体的聚合物胶囊壁,得到光子晶体胶囊墨水。本发明采用微流控技术能够实现对光子晶体的高效包裹,得到大小均一可控的光子晶体胶囊,通过界面聚合产生的聚合物薄膜可以实现对光子晶体的保护,使得光子晶体性能稳定,延长其使用寿命。
附图说明
图1为实施例1制备的光子晶体胶囊的偏光显微镜下光学图像;
图2为实施例1制备的光子晶体胶囊反射光谱图;
图3为实施例1制备的光子晶体胶囊的水分散液在不同时间的颜色变化;
图4为实施例2制备的光子晶体胶囊的偏光显微镜下光学图像;
图5为实施例2制备的光子晶体胶囊反射光谱图;
图6为实施例2制备的光子晶体胶囊的水分散液在不同时间的颜色变化;
图7为实施例3制备的光子晶体胶囊的偏光显微镜下光学图像;
图8为实施例3制备的光子晶体胶囊反射光谱图;
图9为实施例3制备的光子晶体胶囊的水分散液在不同时间的颜色变化。
具体实施方式
本发明提供了一种具有电光响应特性的光子晶体胶囊的制备方法,其特征在于,包括以下步骤:
采用微流控技术,以由一维光子晶体和油溶性聚合前体组成的油相为分散相,以由表面活性剂和超纯水组成的水相为连续相,以由水溶性聚合前体、表面活性剂和超纯水组成的水相为收集相,油相中的油溶性聚合前体和收集相中的水溶性聚合前体在油水交界处发生界面聚合生成包裹光子晶体的聚合物胶囊壁,得到光子晶体胶囊墨水。
本发明采用微流控技术进行光子晶体胶囊的制备,其中,以由一维光子晶体和油溶性聚合前体组成的油相为分散相。
在本发明中,所述一维光子晶体的反射波长位于400~800nm,优选为500~700nm。
所述一维光子晶体按照如下方法进行制备:
将向列相液晶与手性掺杂剂混合,得到一维光子晶体。
其中,所述手性向列相液晶选自型号为bhr-59001、5cb或e7的手性向列相液晶,优选为型号为bhr-59001或5cb的手性向列相液晶;所述手性掺杂剂选自型号为s-811、r811、cb15或r5011的手性向列相液晶,优选为,型号为s-811或cb15的手性向列相液晶所述手性掺杂剂占所述一维光子晶体的质量百分比为20.2wt%~28.6wt%,优选为23.0wt%~25.0wt%。
在所述分散相中,所述油溶性聚合前体为四乙烯五胺(tepa),所述分散相中油溶性聚合前体的质量百分比为1%~5%,优选为3%~4%。
在本发明中,所述分散相按照如下方法进行制备:
将一维光子晶体和油溶性聚合前体混合后加热至80~120℃,电磁搅拌,直到二者完全共溶,形成均匀稳定的分散相。
本发明以由表面活性剂和超纯水组成的水相为连续相。
其中,所述连续相中的表面活性剂选自十二烷基硫酸钠或十二烷基苯磺酸钠,所述连续相中的表面活性剂的质量百分比为1%~5%,优选为3%~4%。
所述连续相按照如下方法进行制备:
将表面活性剂与超纯水混合后超声,直到二者完全共溶,形成均匀稳定的连续相。
本发明以由水溶性聚合前体、表面活性剂和超纯水组成的水相为收集相。
所述收集相中的表面活性剂选自十二烷基硫酸钠或十二烷基苯磺酸钠,所述收集相中表面活性剂的质量百分比为1%~5%,优选为3%~4%;所述收集相中的水溶性聚合前体为甲苯二异氰酸酯(tdi),所述收集相中的水溶性聚合前体的质量百分比为1%~5%,优选为3%~4%。
在本发明中,所述收集相按照如下方法进行制备:
将表面活性剂、tdi和超纯水混合后超声,直到三者完全共溶,形成均匀稳定的收集相。
在本发明中,所述分散相、连续相和收集相的制备顺序没有特殊限制。
得到分散相、连续相和收集相后,本发明采用微流控技术进行光子晶体胶囊的制备,其中,本发明对所述微流控技术中所用的微流控系统并没有特殊限制,能够实现油相中的油溶性聚合前体和收集相中的水溶性聚合前体在油水交界处发生界面聚合生成包裹光子晶体的聚合物胶囊壁即可。
在所述微流控系统的微流控芯片中,所述分散相的流速为1~4微升每分钟,优选为2~3微升每分钟,所述连续相的流速为40~400微升每分钟,优选为50~300微升每分钟,所述收集相在收集瓶中的体积为2~40毫升。最终,可以通过微流控技术得到大小均一可控的光子晶体胶囊。
本发明还提供了一种上述制备方法制备得到的具有电光响应特性的光子晶体胶囊,包括一维光子晶体以及包覆于所述一维光子晶体表面的聚合物胶囊壁,所述光子晶体胶囊的半径为40~150微米,优选为60~120微米,反射波长位于400~800nm,优选为500~700nm。
本发明还提供了一种上述制备方法制备得到的具有电光响应特性的光子晶体胶囊在显示、防伪或信息记录中的应用。
在本发明中,将制备得到的光子晶体胶囊分散于水中,得到光子晶体胶囊墨水。
将光子晶体胶囊墨水灌装至两片导电玻璃之间,用树脂封装导电玻璃,在上下两片导电玻璃之间施加电压,位于导电玻璃之间的光子晶体胶囊墨水具有电光响应的特征,可用于显示中。
另外,本发明制备的光子晶体胶囊还可以用于防伪中,通过改变环境的温度等条件,例如,可以加热到液晶相变温度50~70℃,具有颜色的胆甾相会变为透明的液体,失去颜色。
本发明采用微流控技术以及界面聚合反应能够实现对光子晶体的高效包裹以及“模板法”制备,得到大小均一可控的光子晶体胶囊,通过界面聚合产生的聚合物薄膜可以实现对光子晶体的保护,使得光子晶体性能稳定一致,延长其使用寿命。
为了进一步理解本发明,下面结合实施例对本发明提供的具有电光响应特性的光子晶体胶囊及其制备方法进行说明,本发明的保护范围不受以下实施例的限制。
实施例1
(1)微通道中油相的制备
所选用的一维光子晶体为型号为bhr-59001的手性向列相液晶和型号为s-811的手性掺杂剂,其中,所述手性掺杂剂占所述一维光子晶体的质量百分比为28.5wt%,油溶性聚合前体为四乙烯五胺(tepa)。其中,光子晶体的反射波长位于460nm,油相中tepa的质量百分比为2%。将二者混合后加热至80℃,电磁搅拌,直到二者完全共溶,形成均匀稳定的油相。
(2)微通道中水相的制备
水相中所选用的表面活性剂为十二烷基硫酸钠。水相中表面活性剂的质量百分比为4%。将tdi与超纯水二者混合后超声30分钟,直到二者完全共溶,形成均匀稳定的水相。
(3)收集相的制备
收集相中所选用的表面活性剂为十二烷基硫酸钠。收集相中表面活性剂的质量百分比为4%,甲苯二异氰酸酯(tdi)的质量百分比为2%,。将表面活性剂、tdi和超纯水混合后超声30分钟,直到三者完全共溶,形成均匀稳定的收集相。
(4)光子晶体胶囊的制备
设定微通道中分散相的流速20微升每分钟,连续相的流速为400微升每分钟,收集瓶中的收集相体积为10毫升,使得到的光子晶体胶囊的半径为75微米。
(5)相关性能测试
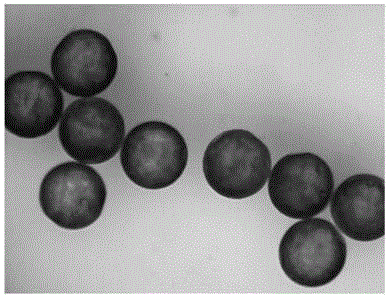
对得到的光子晶体胶囊进行偏光显微镜下观察,结果见图1,图1为实施例1制备的光子晶体胶囊的偏光显微镜下光学图像。
对得到的光子晶体胶囊进行反射光谱测定,结果见图2,图2为实施例1制备的光子晶体胶囊反射光谱图,从图2中可以看出,所述蓝色光子晶体胶囊的反射波长为460nm。
将得到的光子晶体胶囊分散于水中,在一天、一个月和三个月进行观察,结果见图3,图3为实施例1制备的光子晶体胶囊的水分散液在不同时间的颜色变化。从图3中可以看出,本发明制备的光子晶体胶囊基本上无损害,颜色也没有变化。
实施例2
(1)微通道中油相的制备
所选用的一维光子晶体为型号为5cb的手性向列相液晶和型号为s-811的手性掺杂剂,其中,所述手性掺杂剂占所述一维光子晶体的质量百分比为26.2wt%,油溶性聚合前体为四乙烯五胺(tepa)。其中,光子晶体的反射波长位于550nm,油相中tepa的质量百分比为3%。将二者混合后加热至80℃,电磁搅拌,直到二者完全共溶,形成均匀稳定的油相。
(2)微通道中水相的制备
水相中所选用的表面活性剂为十二烷基硫酸钠。水相中表面活性剂的质量百分比为5%。将tdi与超纯水二者混合后超声30分钟,直到二者完全共溶,形成均匀稳定的水相。
(3)收集相的制备
收集相中所选用的表面活性剂为十二烷基硫酸钠。收集相中表面活性剂的质量百分比为5%,甲苯二异氰酸酯(tdi)的质量百分比为3%,。将表面活性剂、tdi和超纯水混合后超声30分钟,直到三者完全共溶,形成均匀稳定的收集相。
(4)光子晶体胶囊的制备
设定微通道中分散相的流速10微升每分钟,连续相的流速为300微升每分钟,收集瓶中的收集相体积为10毫升,使得到的光子晶体胶囊的半径为90微米。
(5)相关性能测试
对得到的光子晶体胶囊进行偏光显微镜下观察,结果见图4,图4为实施例2制备的光子晶体胶囊的偏光显微镜下光学图像。
对得到的光子晶体胶囊进行反射光谱测定,结果见图5,图5为实施例2制备的光子晶体胶囊反射光谱图,从图5中可以看出,所述绿色光子晶体胶囊的反射波长为550nm。
将得到的光子晶体胶囊分散于水中,在一天、一个月和三个月进行观察,结果见图6,图6为实施例2制备的光子晶体胶囊的水分散液在不同时间的颜色变化。从图6中可以看出,本发明制备的光子晶体胶囊基本上无损害,颜色也没有变化。
实施例3
(1)微通道中油相的制备
所选用的一维光子晶体为型号为bhr-59001的手性向列相液晶和型号为cb15的手性掺杂剂,其中,所述手性掺杂剂占所述一维光子晶体的质量百分比为25.1wt%,油溶性聚合前体为四乙烯五胺(tepa)。其中,光子晶体的反射波长位于750nm,油相中tepa的质量百分比为4%。将二者混合后加热至80℃,电磁搅拌,直到二者完全共溶,形成均匀稳定的油相。
(2)微通道中水相的制备
水相中所选用的表面活性剂为十二烷基硫酸钠。水相中表面活性剂的质量百分比为5%。将tdi与超纯水二者混合后超声30分钟,直到二者完全共溶,形成均匀稳定的水相。
(3)收集相的制备
收集相中所选用的表面活性剂为十二烷基硫酸钠。收集相中表面活性剂的质量百分比为5%,甲苯二异氰酸酯(tdi)的质量百分比为4%,。将表面活性剂、tdi和超纯水混合后超声30分钟,直到三者完全共溶,形成均匀稳定的收集相。
(4)光子晶体胶囊的制备
设定微通道中分散相的流速5微升每分钟,连续相的流速为100微升每分钟,收集瓶中的收集相体积为10毫升,使得到的光子晶体胶囊的半径为120微米。
(5)相关性能测试
对得到的光子晶体胶囊进行偏光显微镜下观察,结果见图7,图7为实施例3制备的光子晶体胶囊的偏光显微镜下光学图像。
对得到的光子晶体胶囊进行反射光谱测定,结果见图8,图8为实施例3制备的光子晶体胶囊反射光谱图,从图8中可以看出,所述红色光子晶体胶囊的反射波长为750nm。
将得到的光子晶体胶囊分散于水中,在一天、一个月和三个月进行观察,结果见图9,图9为实施例3制备的光子晶体胶囊的水分散液在不同时间的颜色变化。从图9中可以看出,本发明制备的光子晶体胶囊基本上无损害,颜色也没有变化。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!