一种硝基苯并异噻唑-吡啶酮染料及其制备方法与流程
本发明涉及一种硝基苯并异噻唑-吡啶酮染料及其制备方法,属于杂环染料合成技术领域。
背景技术:
杂环分散染料相比于传统的苯环偶氮分散染料色泽更鲜艳、发色强度更高。它们不但在纺织工业染料着色剂方面有着广泛的应用,而且在电子工业如非线性光学器件、可视传感器和染料敏化电池方面也发挥着重要作用。其中以吡啶-2,6-二酮及其衍生物作为偶合组分所合成的杂环染料色泽非常鲜艳而且各种牢度性能优异。
研究表明,以3-氨基-5-硝基-苯并异噻唑为重氮组分,吡啶-2,6-二酮为偶合组分所合成的硝基苯并异噻唑-吡啶酮杂环染料,在酸性环境的诱导下由于活泼质子的迁移存在偶氮-腙式互变异构现象,在中性条件下以偶氮结构存在,在酸性条件下转化为腙式结构。这种现象使其在不同极性的溶剂诱导下,会呈现出鲜艳的黄、红和蓝色。然而,染料对纤维进行染色作用时,通常是在酸性条件下,这就意味着染色过程中染料由于发生偶氮-腙式互变异构现象导致染色的稳定性下降。导致这种现象最可能的原因是:偶合组分吡啶-2,6-二酮存在一个活泼的羟质子,该质子可在酸性条件下发生偶氮部分和吡啶酮环的可逆迁移(如以下的反应式所示)。目前文献中多利用过渡金属与偶氮染料形成配合物的方式[(a)you,w.;zhu,h.y.;huang,w.;hu,b.;fan,y.;you,x.z.,daltontrans.2010,39,7876-7880;(b)chen,x.c.;tao,t.;wang,y.g.;peng,y.x.;huang,w.;qian,h.f.,daltontrans.2012,41,11107-11115;(c)zhao,x.l.;qian,h.f.;huang,w.,dyespigments2018,149,796-803.],拔除活泼质子,以消除杂环染料偶氮-腙式互变异构现象。然而,由此形成的庞大染料分子结构不利于与纤维的进一步作用,反而降低染料染色的各种牢度参数。
技术实现要素:
针对上述问题,本发明提供了一种硝基苯并异噻唑-吡啶酮染料及其制备方法,本发明通过官能团转化的方式实现吡啶-2,6-二酮酸性的羟基被碱性的胺基取代,通过此方法拔除活泼质子,能够提高吡啶酮基杂环染料的酸稳定性,从而消除由于互变异构现象导致的染色不稳定的问题。
本发明的第一个目的是提供一种硝基苯并异噻唑-吡啶酮染料,所述硝基苯并异噻唑-吡啶酮染料的分子结构式如式i所示:
式中,r1为-[ch2]n-o-ch3,n为3-8;r2选自氢、c1~8烷基、羟烷基、烷氧基烷基、苯基。
本发明的第二个目的是提供一种制备上述硝基苯并异噻唑-吡啶酮染料的方法,所述方法通过以下反应式进行:
式中,r1为-[ch2]n-o-ch3,n为3-8;r2选自氢、c1~8烷基、羟烷基、烷氧基烷基、苯基。
在本发明的一种实施方式中,所述制备上述硝基苯并异噻唑-吡啶酮染料的方法,包括如下步骤:
(1)将1-n-r2-3-氰基-4-甲基-6-羟基-2-吡啶酮溶解于三氯氧磷中并加热,反应完毕后,洗涤反应产物,然后利用二氯甲烷溶解产物,再进行分离,分离得到的有机相干燥过滤,收集滤液并除去溶剂,最后经纯化得到化合物(i);
(2)将化合物(i)溶解在无水乙醇中,加入nh2-(ch2)n-o-ch3(n为3-8)并加热,反应完毕后除去反应产物中的溶剂,最后经纯化得到化合物(ii);
(3)将3-氨基-5-硝基-2,1-苯并异噻唑用浓硫酸和冰醋酸溶解,得到混合液,将亚硝酸钠溶于水后加入到所得混合液中并搅拌,得到重氮盐;将化合物(ii)溶解于甲醇-水的混合溶液中,得到化合物(ii)的混合溶液;
(4)将所得重氮盐加到化合物(ii)的混合溶液中并搅拌,反应完毕后,将反应母液加热、陈化,然后过滤,用水洗涤得到粗品;
(5)将粗品用有机溶剂溶解,然后加热回流,冷却至室温析出晶体,得到最终产物。
在本发明的一种实施方式中,步骤(1)中所述1-n-r2-3-氰基-4-甲基-6-羟基-2-吡啶酮与所述三氯氧磷摩尔比为1:2~1:2.2,所述加热温度为80-100℃。
在本发明的一种实施方式中,步骤(1)中所述洗涤为采用冰水混合物对反应产物洗涤。
在本发明的一种实施方式中,步骤(1)中所述分离的方式为转移到分液漏斗中萃取。
在本发明的一种实施方式中,步骤(1)中所述干燥为采用无水硫酸钠进行干燥并静置5-10分钟,利用旋转蒸发仪除去溶剂。
在本发明的一种实施方式中,步骤(2)中所述化合物(i)与所述nh2-(ch2)n-o-ch3的摩尔比为1:2~1:2.2,所述加热温度为80-100℃。
在本发明的一种实施方式中,步骤(3)中所述3-氨基-5-硝基-2,1-苯并异噻唑与所述亚硝酸钠与所述化合物(ii)的摩尔比为1:1.1:1。
在本发明的一种实施方式中,步骤(3)中所述冰醋酸与所述浓硫酸的体积比为1:1~1:1.5,所述搅拌时间为20-40分钟,所述甲醇-水的混合溶剂中甲醇与水体积比为2:1。
在本发明的一种实施方式中,步骤(4)中所述重氮盐和化合物(ii)混合的摩尔比为1:1~1:1.2。
在本发明的一种实施方式中,步骤(4)中所述加热温度为45-65℃,所述陈化时间为10-20分钟。
在本发明的一种实施方式中,步骤(5)中所述有机溶剂为三氯甲烷-乙醇混合溶剂、三氯甲烷-甲醇混合溶剂、二氯甲烷-乙醇混合溶剂中任一种。
在本发明的一种实施方式中,所述三氯甲烷-乙醇混合溶剂中三氯甲烷与乙醇体积比为1:1,所述三氯甲烷-甲醇混合溶剂中三氯甲烷与甲醇体积比为1:1,所述二氯甲烷-乙醇混合溶剂中二氯甲烷与乙醇体积比为1:1。
在本发明的一种实施方式中,步骤(5)中所述加热温度为50-70℃,回流时间为30-60分钟。
在本发明的一种实施方式中,步骤(1)和步骤(2)中所述纯化为利用色谱柱进行纯化。
在本发明的一种实施方式中,步骤(1)中所述柱色谱法使用硅胶填充剂作为固定相,使用乙酸乙酯-石油醚洗脱剂作为流动相,进行柱色谱法纯化,其中乙酸乙酯与石油醚的体积比为1:1;步骤(2)中所述柱色谱法使用硅胶填充剂作为固定相,使用二氯甲烷-甲醇洗脱剂作为流动相,进行柱色谱法纯化,其中二氯甲烷与甲醇体积比为80:1。
本发明的第三个目的是提供一种纺织工业中染料着色剂,所述染料着色剂中含有上述硝基苯并异噻唑-吡啶酮染料。
本发明的第四个目的是提供上述硝基苯并异噻唑-吡啶酮染料在非线性光学器件、可视传感器和染料敏化电池中的应用。
本发明的有益效果:
(1)本发明通过官能团转化的方式采用碱性胺基对吡啶酮的羟基进行改性,然后以改性后的吡啶酮为偶合组分,以3-氨基-5-硝基-2,1-苯并异噻唑为重氮组分,合成了对酸性环境稳定的双杂环偶氮染料。在不同的酸环境诱导下,通过官能团转化合成的染料分子由于没有活泼羟基质子的存在,不存在偶氮-腙式互变异构现象,具有好的酸稳定性。
(2)本发明反应条件易控,操作方便,产物收率高。
附图说明
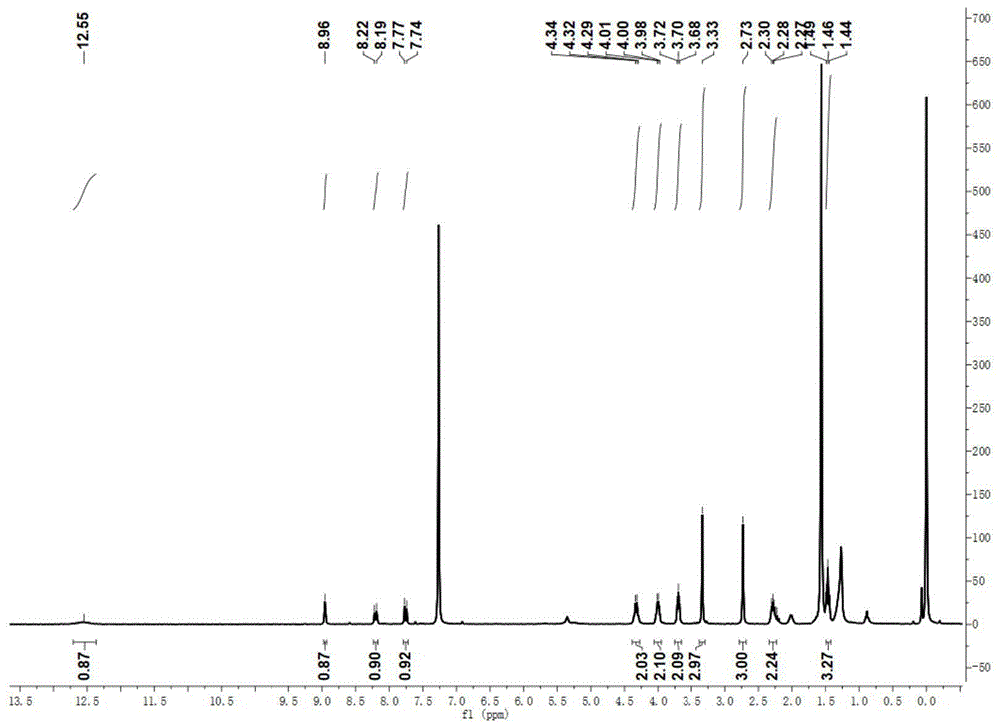
图1为实施例1中制备的化合物i-1的核磁氢谱图。
图2为实施例1中制备的化合物i-1的质谱图。
图3为实施例1中制备的化合物i-1在不同酸性环境下的的uv-vis吸收光谱图。
图4为实施例2中制备的化合物i-2的核磁氢谱图。
图5为实施例2中制备的化合物i-2的质谱图。
图6为实施例2中制备的化合物i-2在不同酸性环境下的的uv-vis吸收光谱图。
具体实施方式
下面结合具体实施例,对本发明作进一步详细阐述。
实施例1
制备化合物i-1:
(1)取10.00mmol1-n-乙基-3-氰基-4-甲基-6-羟基-2-吡啶酮溶解于21.60mmol三氯氧磷中并80℃加热,通过tlc监测反应完毕后,将反应产物用冰水混合物洗涤,然后用二氯甲烷充分溶解并转移到分液漏斗中进行萃取,将分离得到的有机相用无水硫酸钠干燥5分钟,然后过滤,收集滤液用旋转蒸发仪除去溶剂得到红色粗品,使用60.00g硅胶填充剂作为固定相,使用500.00ml乙酸乙酯-石油醚(其中乙酸乙酯与石油醚体积比为1:1)洗脱剂作为流动相对粗品进行柱色谱法纯化,得到化合物(i-1),产率为84%;
(2)取5.00mmol化合物(i-1)溶解在25.00ml无水乙醇中,加入11.00mmol3-甲氧基丙胺并90℃加热,通过tlc监测反应完毕后,用旋转蒸发仪除去产物中的溶剂得到粗品,使用40.00g硅胶填充剂作为固定相,使用300.00ml二氯甲烷-甲醇(其中二氯甲烷与甲醇体积比为80:1)洗脱剂作为流动相对粗品进行柱色谱法纯化,得到化合物(ii-1),产率为78%;
(3)取10.00mmol3-氨基-5-硝基-2,1-苯并异噻唑用5.00ml质量分数98%的浓硫酸和5.00ml冰醋酸溶解,得到混合液,取11.00mmol亚硝酸钠用5.00ml去离子水溶解后加入到所得混合液中并搅拌30分钟,得到重氮盐;取10.00mmol化合物(ii-1)用60.00ml甲醇-水(其中甲醇与水体积比为2:1)的混合溶剂溶解,得到化合物(ii-1)的混合溶液;
(4)将所得重氮盐全部加到化合物(ii-1)的混合溶液中并搅拌,通过tlc监测反应完毕后,将反应母液55℃加热、陈化10分钟,然后过滤,用水洗涤得到粗品;
(5)将粗品用20.00ml三氯甲烷-乙醇(其中三氯甲烷与乙醇体积比为1:1)混合溶剂溶解,然后60℃加热回流45分钟后,冷却至室温析出晶体,得到化合物i-1,产率为82%。
上述制备过程如下:
对本实施中制备的化合物进行核磁测试,图1为实施例1中制备的化合物i-1的核磁氢谱图,由化合物i-1的氢核磁共振谱图能够证明获得了终产品。
1hnmr(300mhz,cdcl3,ppm):δ=12.55(s,1h),8.96(s,1h),8.21(d,j=9.7hz,1h),7.76(d,j=9.7hz,1h),4.39-4.27(m,2h),4.06-3.95(m,2h),3.70(t,j=5.3hz,2h),3.33(s,3h),2.73(s,3h),2.28(dd,j=11.2,5.6hz,2h),1.46(t,j=7.0hz,3h).negativeesi–msinmethanol:(m/z)=454.15(100%),[m-h]-。
图2为本实施制备的化合物i-1的esi-ms图,由图2能够看出在甲醇溶剂中使用负离子模式,化合物的分子离子峰以100%的丰度出现,说明该方法分析杂环吡啶酮染料的分子结构是有效的。
酸稳定测试:
配置浓度为3.0×10-5mol/l化合物i-1的甲醇溶液、0.1mol/l醋酸的甲醇溶液。首先,测试化合物i-1的甲醇溶液的uv-vis吸收光谱(ph=6.77的吸收谱线),之后向化合物i-1的甲醇溶液中滴加0.1mol/l醋酸的甲醇溶液,并测试其酸碱度和uv-vis吸收光谱(ph=1.35,3.47,6.77的吸收谱线),如图3所示。由图3可知,经过官能团转化方式合成的化合物i-1其最大吸收波长位于549nm处,且不随外界酸环境的改变而改变,证明该染料有高的酸稳定性。
实施例2
制备化合物i-2:
(1)取10.00mmol1-n-异丙氧基丙基-3-氰基-4-甲基-6-羟基-2-吡啶酮溶解于21.60mmol三氯氧磷中并80℃加热,通过tlc监测反应完毕后,将反应产物用冰水混合物洗涤,然后用二氯甲烷充分溶解并转移到分液漏斗中进行萃取,将分离得到的有机相用无水硫酸钠干燥5分钟,然后过滤,收集滤液用旋转蒸发仪除去溶剂得到红色粗品,使用60.00g硅胶填充剂作为固定相,使用500.00ml乙酸乙酯-石油醚(其中乙酸乙酯与石油醚体积比为1:1)洗脱剂作为流动相对粗品进行柱色谱法纯化,得到化合物(i-2),产率为90%;
(2)取5.00mmol化合物(i-2)溶解在25.00ml无水乙醇中,加入11.00mmol3-甲氧基丙胺并90℃加热,通过tlc监测反应完毕后,用旋转蒸发仪除去产物中的溶剂得到粗品,使用40.00g硅胶填充剂作为固定相,使用300.00ml二氯甲烷-甲醇(其中二氯甲烷与甲醇体积比为80:1)洗脱剂作为流动相对粗品进行柱色谱法纯化,得到化合物(ii-2),产率为80%;
(3)取10.00mmol3-氨基-5-硝基-2,1-苯并异噻唑用5.00ml质量分数98%的浓硫酸和5.00ml冰醋酸溶解,得到混合液,取11.00mmol亚硝酸钠用5.00ml去离子水溶解后加入到所得混合液中并搅拌30分钟,得到重氮盐;取10.00mmol化合物(ii-2)用60.00ml甲醇-水(其中甲醇与水体积比为2:1)的混合溶剂溶解,得到化合物(ii-2)的混合溶液;
(4)将所得重氮盐全部加到化合物(ii-2)的混合溶液中并搅拌,通过tlc监测反应完毕后,将反应母液55℃加热、陈化10分钟,然后过滤,用水洗涤得到粗品;
(5)将粗品用20.00ml三氯甲烷-乙醇(其中三氯甲烷与乙醇体积比为1:1)混合溶剂溶解,然后60℃加热回流45分钟后,冷却至室温析出晶体,得到化合物i-2,产率为85%。
上述制备过程如下:
对本实施中制备的化合物进行核磁测试,图4为实施例2中制备的化合物i-2的核磁氢谱图,由化合物i-2的氢核磁共振谱图能够证明获得了终产品。
1hnmr(400mhz,cdcl3,ppm):δ=12.69(s,1h),8.98(d,j=1.7hz,1h),8.20(dd,j=9.7,2.3hz,1h),7.77-7.73(m,1h),4.44-4.37(m,2h),4.06(dd,j=11.2,6.5hz,2h),3.69-3.64(m,2h),3.58-3.47(m,3h),3.30(s,3h),2.71(s,3h),2.24(dd,j=11.6,6.0hz,2h),2.03(dd,j=13.1,7.4hz,2h),1.14(d,j=6.1hz,6h).negativeesi–msinmethanol:(m/z)=526.25(100%),[m-h]-。
图5为本实施制备的化合物i-2的esi-ms图,由图5能够看出在甲醇溶剂中使用负离子模式,化合物的分子离子峰以100%的丰度出现,说明该方法分析杂环吡啶酮染料的分子结构是有效的。
酸稳定测试:
配置浓度为3.0×10-5mol/l化合物i-2的甲醇溶液、0.1mol/l醋酸的甲醇溶液。首先,测试化合物i-2的甲醇溶液的uv-vis吸收光谱(ph=7.01的吸收谱线),之后向化合物i-2的甲醇溶液中滴加0.1mol/l醋酸的甲醇溶液,并测试其酸碱度和uv-vis吸收光谱(ph=1.25,4.52,7.01的吸收谱线),如图6所示。由图6可知,经过官能团转化方式合成的化合物i-2其最大吸收波长位于555nm处,且不随外界酸环境的改变而改变,证明该染料有高的酸稳定性。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!